Медичні загадки: рідкісні ендокринні й метаболічні синдроми
Мозаїку симптомів рідкісних ендокринних патологій збирала Владислава Зінченко
У світі налічується понад 1500 рідкісних (орфанних) ендокринних і метаболічних захворювань. Рідкісними вважаються хвороби, що спостерігаються з частотою ≤1:2000. Розмиті симптоми, нетипова клінічна картина, складна діагностика, що потребує глибоких медичних знань і лікарської інтуїції, – ось типові характеристики таких патологій. За даними М.Д. Тронька та співавт. (2020), в Україні офіційно затверджено 302 нозології, які віднесено до рідкісних захворювань, серед них 61 рідкісна ендокринна хвороба (в тому числі розлади харчування та порушення обміну речовин), а також уроджені вади розвитку, хромосомні аномалії та рідкісні новоутворення, у виборі тактики ведення котрих безпосередньо задіяний ендокринолог. Розгляньмо деякі з них.
Синдром Ларона (гіпофізарна карликовість) – спадкова хвороба з переважно автосомно-рецесивним шляхом передачі, що характеризується низьким зростом (менш ніж 130 см) і нечутливістю периферичних тканин до дії гормону росту, зумовленою дефектом гена рецептора соматотропного гормону (ген GHR). При народженні зріст дитини нормальний, сповільнюється він із 1-го року життя.

У пацієнтів спостерігаються надмірна маса тіла, затримка статевого розвитку, м’язова гіпотонія, фенотипово – виступає ніс і западає перенісся, блакитні склери, тонке й ламке волосся, аномалії зубів, короткі кінцівки.
Близько третини хворих на синдром Ларона (~100 із 350) проживають у південних районах Еквадору (фото 1). Позитивний аспект порушення: в пацієнтів із синдромом Ларона знижується ризик появи цукрового діабету 2 типу й онкопатології.
Терапія гормоном росту неефективна й не застосовується. В Європі та США використовують рекомбінантний інсуліноподібний фактор росту-1.
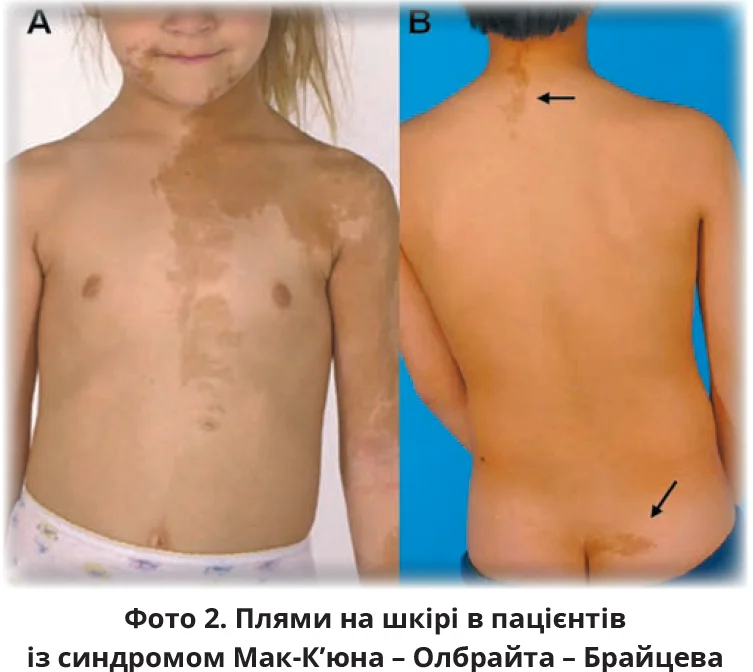
Синдром Мак-К’юна – Олбрайта – Брайцева є рідкісною генетичною патологією, зумовленою соматичною мутацією гена GNAS, що виникає
до 10-го тижня внутрішньоутробного розвитку. Клінічно проявляється множинними асиметричними плямами кольору кави з молоком на шкірі (з нерівними зубчатими краями, фото 2), поліосальною фіброзною дисплазією кісток і гіперфункцією ендокринних залоз (передчасне статеве дозрівання).
Також можуть бути уражені й інші ендокринні залози: щитоподібна й паращитоподібні, кора наднирників, яєчники та ін., що спричиняє гормональну бурю в організмі дитини. Статеве дозрівання починається від 6 місяців до 7 років, середній вік появи перших ознак передчасного статевого дозрівання, зокрема менструальноподібних виділень,
у дівчаток – 2-3 роки.
Зазвичай синдром дебютує в ранньому віці (приблизно 1-2 роки), є поширенішим серед дівчат. Частота варіює від 1 випадку на 100 тис. населення до 1 – на 1 млн загальної популяції.
Протягом останніх років клінічна тріада дещо стерлася, синдром має мозаїчний характер, тому патологія дуже складна в діагностиці, що корелює з високим ризиком інвалідизації (втрата рухової функції нижніх кінцівок, часті переломи, тяжкі деформації суглобових поверхонь, деформація проксимального відділу стегнової кістки кульшового суглоба за типом «палиці пастуха» чи «бумеранга», іноді з укороченням кінцівки до 10 см).
На сьогодні специфічного лікування цього синдрому не існує. Такі пацієнти потребують спостереження команди фахівців – педіатра, ендокринолога, хірурга, ортопеда, гінеколога, невролога, офтальмолога та ін. На щастя, хвороба не успадковується.

Синдром кленового сиропу – спадкове порушення з групи органічних ацидемій, спричинене дефіцитом дегідрогенази кетокислот із розгалуженим ланцюгом і порушенням метаболізму амінокислот лейцину, ізолейцину, валіну (внаслідок чого вони накопичуються в організмі). Має автосомно-рецесивний тип наслідування. Частота виявлення – 1 випадок на 120-300 тис. новонароджених. У перші 2 тижні життя в малюків спостерігаються відмова від їжі, часті зригування, блювання. Сечі пацієнтів із цим захворюванням притаманний запах кленового сиропу. У разі відсутності лікування з’являються судоми окремих груп м’язів, посилюється м’язовий тонус, виникають порушення дихання, затримка психологічного й фізичного розвитку. Лікування синдрому передбачає спеціальну дієту. Використовують суміші амінокислот і гідролізати, що не містять амінокислот із розгалуженим ланцюгом.
Синдром Прадера – Віллі є рідкісним генетично зумовленим захворюванням, у разі якого сім генів (або їхні частини) на батьківській хромосомі 15q11.2-q13 видалені або не здатні повноцінно функціонувати. Припускають, що клінічна симптоматика пов’язана з дисфункцією гіпоталамуса, хоча чітких доказів цієї гіпотези досі не отримали. Частота синдрому – 1 випадок на 10-25 тис. новонароджених (без гендерних відмінностей). Наразі у світі таких пацієнтів близько 400 тис.
Існують діагностичні маркери, що дають змогу запідозрити патологію ще до народження дитини.
У внутрішньоутробному періоді синдром проявляється зниженням активності руху плода, ненормальним його положенням, багатоводдям. У дитячому й підлітковому віці у хворих спостерігаються низький зріст, крайній ступінь ожиріння, небезпечний для життя (фото 4), гіпогонадизм, косоокість, затримка розумового розвитку та статевого дозрівання (недорозвиненість статевих органів), аномальна гнучкість, денна сонливість, слабкість м’язів, часто безпліддя.
Візуально відзначаються мигдалеподібні очі з вузькими опущеними вниз повіками, вузький лоб, стриї, диспропорція тіла (маленькі руки й ноги з вузькими пальцями), синці (пацієнти мають схильність до їх появи), подряпини й інші ушкодження шкіри (прояв дерматіломанії), сколіоз/кіфоз.
Спостерігається поліфагія, хворі не можуть контролювати відчуття голоду, вони можуть шукати їжу в смітнику, красти її, навіть споживати корм для тварин. Окрім того, пацієнтам притаманні зниження психомоторного розвитку, порушення мовлення, обмежений словниковий запас. Підвищується ризик розвитку цукрового діабету 2 типу, синдрому обструктивного апное сну. Збільшується концентрація греліну в крові, зменшується – соматоліберину.
У разі підозри на синдром Прадера – Віллі рекомендуються генетичні тести. Специфічна терапія відсутня. Дітям призначаються щоденні ін’єкції рекомбінантного гормону росту. Соматотропін (соматотропний гормон гіпофізу) сприяє збільшенню м’язової маси та зниженню апетиту хворих. Дуже важливими є фізіотерапія, корекція маси тіла.
Вперше синдром Прадера – Віллі був описаний у британських ЗМІ в липні 2007 р.: телевізійний канал Channel 4 присвятив програму під назвою Can’t Stop Eating («Не можу припинити їсти») особливостям життя Джо й Тамари, що страждають від хвороби.

Алкаптонурія – спадкова хвороба (автосомно-рецесивний тип наслідування), що характеризується порушенням обміну тирозину, екскрецією із сечею гомогентизинової кислоти. Виникає внаслідок мутації гена, що кодує оксидазу гомогентизинової кислоти, розташованого на 3-й хромосомі. Частіше трапляється серед хлопчиків.
Першим клінічним випадком алкаптонурії, датованим іще 1500 р. до н. е., й пацієнтом із цією ензимопатією вважається єгипетська мумія Harwa (F. Stenn et al., 1977). Поширеність хвороби коливається від 1:100 тис. до 1:250 тис. Значну кількість випадків зафіксовано в Словаччині, Домініканській Республіці, Індії, Йорданії.
Через дефект ферменту оксидази процес перетворення гомогентизинової кислоти порушується; вона перетворюється на бензохінонацетат або алкаптон (хініновий поліфенол), який виводиться нирками. Алкаптон накопичується в сполучній тканині, що зумовлює її підвищену крихкість і потемніння.
Рання клінічна ознака – виділення в дитини сечі, яка швидко темніє при перебуванні в ємності на повітрі, при підігріванні, залуженні («хвороба чорної сечі»). Хворі мають схильність до розвитку сечокам’яної хвороби, пієлонефриту, гіпотиреозу.
У дорослому віці (після 30 років) виникають ураження опорно-рухового апарату: кульшових, колінних, плечових суглобів; синовіт і деструктивні зміни хряща суглобів; біль у різних відділах хребта; накопичення гомогентизинової кислоти в сухожилках, зв’язках, що супроводжується кальцифікацією й запаленням; на рентгенограмах – кальцифікація міжхребцевих дисків; зміни аортального/мітрального клапанів.
Візуально можуть виявлятися зміна кольору вушних раковин на сірий чи блакитний, зміна кольору долонь, ділянок під пахвами, носогубних складок, пігментація склер (фото 5).
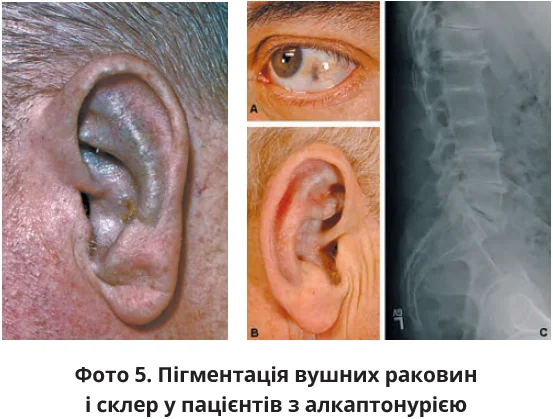
Діагностика ґрунтується на кількісному визначенні бензохіноноцтової та гомогентизинової кислот у сечі за допомогою рідинної хроматографії чи ферментативної спектрофотометрії. Простіший, але й менш точний спосіб – оцінити колір сечі після перебування її на повітрі протягом 12-24 годин: через окислення алкаптону вона стає бурою чи чорною (за умови лужного рН сечі).
Специфічного лікування не існує. Призначають симптоматичну терапію й високі дози аскорбінової кислоти, низькобілкову дієту. Перспективним лікарським засобом вважають нітизинон.
Акромегалія й гіпофізарний гігантизм досить прості в діагностиці з огляду на характерний зовнішній вигляд пацієнтів, але через
фінансові й інші причини складні в лікуванні. Вони зумовлені надмірною секрецією гормону росту (гіперсоматотропізмом) унаслідок (у більшості випадків) аденоми передньої частки гіпофізу.
До закриття епіфізарних зон росту кісток це призводить до розвитку гігантизму: швидкість росту скелета та зріст збільшуються, кістки практично не деформуються, спостерігається затримка статевого розвитку або гіпогонадотропний гіпогонадизм із характерним євнухоподібним типом будови тіла (висока струнка статура, довгі кінцівки). Якщо ж надмірна секреція гормону росту відбувається після припинення росту організму, це спричиняє акромегалію. За відсутності лікування гігантизм може поєднуватися з акромегалією.
Поширеність акромегалії становить 4600 випадків на 1 млн населення. Середній вік установлення діагнозу – 40 років у чоловіків, 45 років – у жінок.
За даними 2020 р., в Україні офіційно зареєстровано 844 пацієнти з акромегалією (експерти припускають, що реальна кількість сягає 2500).

Акромегалія супроводжується непропорційним збільшенням (розширенням і потовщенням) кистей, підошов, черепа (м’яких тканин, носа, підборіддя, виличних кісток, надбрівних дуг) тощо (фото 6). Хворі постійно змінюють розмір каблучок, рукавичок, взуття. Шкіра потовщена, часто темного кольору, нижня щелепа та язик збільшені, міжзубні проміжки розширені. Грудна клітка має бочкоподібну форму. Відзначаються огрубіння голосу, зниження зору.
Також відзначаються посилення обміну речовин, функції сальних залоз, гіпергідроз, синдром зап’ясткового каналу, дегенеративні зміни суглобів, артеріальна гіпертензія, цукровий діабет, галакторея та/або гіперпролактинемія, гіперкальційурія, синдром обструктивного апное сну, головний біль, виражена слабкість; у 2-3 рази збільшується ризик поліпозу й раку товстого кишківника. Приблизно в третини чоловіків з акромегалією діагностується еректильна дисфункція, практично в усіх жінок – порушення менструального циклу, зокрема аменорея.
Зазвичай діагностика хвороби ґрунтується на виявленні характерних клінічних ознак, оцінці концентрації інсуліноподібного фактора росту-1, гормону росту, даних комп’ютерної/магнітно-резонансної томографії. Призначають хірургічне лікування, або медикаментозну терапію (октреотид/ланреотид/пасиреотид; пегвісомант; бромокриптин або каберголін), або променеву терапію.
Акромегалія пов’язана з високим рівнем смертності внаслідок злоякісних новоутворень, кардіоваскулярних і респіраторних порушень. За умови адекватного контролю гормональних показників рівень смертності не перевищує такий у загальній популяції.
Синдром Рассела – Сільвера є рідкісним генетичним порушенням, що характеризується затримкою внутрішньоутробного розвитку й постнатальною низькорослістю. Спричинений аномаліями на рівні 7-ї й 11-ї хромосом. Частота синдрому – 1:30 тис. (за деякими даними, 1:100 тис.).
У новонароджених із захворюванням спостерігаються труднощі вигодовування. Візуально в пацієнтів виступає лоб, відзначаються «трикутне» обличчя, псевдогідроцефалія, опущені кути рота («рот коропа»), вузьке підборіддя, мікрогнатія, вкорочені й зігнуті пальці рук, синдактилія, асиметрія тіла, відставання маси тіла від норми, аномалії зовнішніх статевих органів (крипторхізм, гіпоплазія статевого члена тощо) та сечовидільної системи, короткі руки, поперековий лордоз, зменшення м’язової маси (фото 7). Виявляються підвищені рівні фолікулостимулювального та лютеїнізувального гормонів, гіпоглікемія.
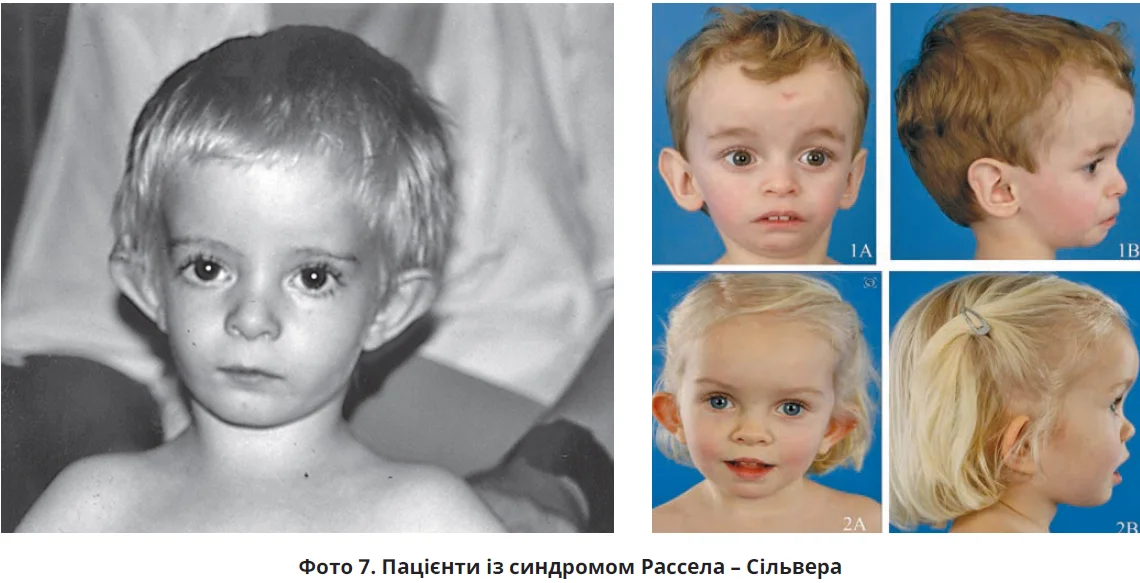
Кінцевий зріст у хворих із синдромом Рассела – Сільвера – на рівні 140-150 см. Інтелект зазвичай збережений. Можлива затримка мовлення. Захворювання зумовлює певні щоденні складнощі, проте не становить безпосередньої загрози для життя. Лікування симптоматичне за участі міждисциплінарної команди.
Широка палітра клінічних порушень, стигматизація, складна та тривала діагностика, високовартісне та/або недоступне лікування – це лише частина труднощів, із якими стикаються люди, що мають орфанні хвороби.
Протягом останніх десятиліть у багатьох країнах були створені або створюються національні реєстри пацієнтів із рідкісними соціально значущими захворюваннями. Такий підхід допомагає планувати стратегію лікування та забезпечувати хворих необхідними препаратами. В Україні наразі існує лише один такий реєстр – пацієнтів зі спінальною м’язовою атрофією.