Фармакологічна модуляція судинного старіння: огляд VascAgeNet. Частина 3
Переклала й адаптувала д-р мед. наук Лариса Стрільчук
Ремоделювання позаклітинного матриксу
Позаклітинний матрикс (ПКМ) являє собою тривимірну архітектурну мережу макромолекул, яка визначає морфологічні та фізичні властивості тканин і органів, а також бере участь у керуванні клітинними процесами. Будь-яка модифікація складу та структури ПКМ, тобто його ремоделювання, має значний вплив на морфологію, механічні й функціональні властивості артеріальної стінки (Wagenseil and Mecham, 2009). Основними компонентами стінки артерій є еластин і колагени. Білі еластичні волокна, які складаються з еластину та мікрофібрил, забезпечують судинам еластичність і пружність, а колагени, передусім колаген 1-го типу, – жорсткість і міцність на розрив. У ході старіння еластин і колаген підлягають змінам і хімічній модифікації, в тому числі через їхній тривалий період напіврозпаду.
Значущою ознакою артерій, які зазнали старіння, є фрагментація еластичних волокон (Duca et al., 2016). Одна з важливих причин цього – механічне руйнування пластинок еластину (рис.) у зв’язку з постійними повторними розтягненнями та розслабленнями, які відбуваються в аорті протягом життя людини. Ці руйнування та навіть розриви призводять до погіршення функцій еластичних волокон і до перенесення механічних навантажень на колагени, що спричиняє розвиток жорсткості та зменшення розтяжності артерій (Greenwald, 2007; Hodis and Zamir, 2009). Фрагментацію еластину підсилюють протеїнази (еластази), причому в результаті цього процесу вивільняються біоактивні пептиди (еластокіни), які модулюють низку біологічних процесів шляхом впливу на комплекс рецепторів до еластину (Tembely et al., 2022). Зокрема, еластокіни модулюють адгезію клітин, хемотаксис, клітинну міграцію, проліферацію, активацію протеїназ, ангіогенез і апоптоз (рис.). Комплекс рецепторів до еластину являє собою гетеротримерний рецептор, який складається з еластинозв’язувального білка, захисного білка катепсину А та нейрамінідази-1 (остання виконує функцію каталітичної субодиниці) (Bennasroune et al., 2019).
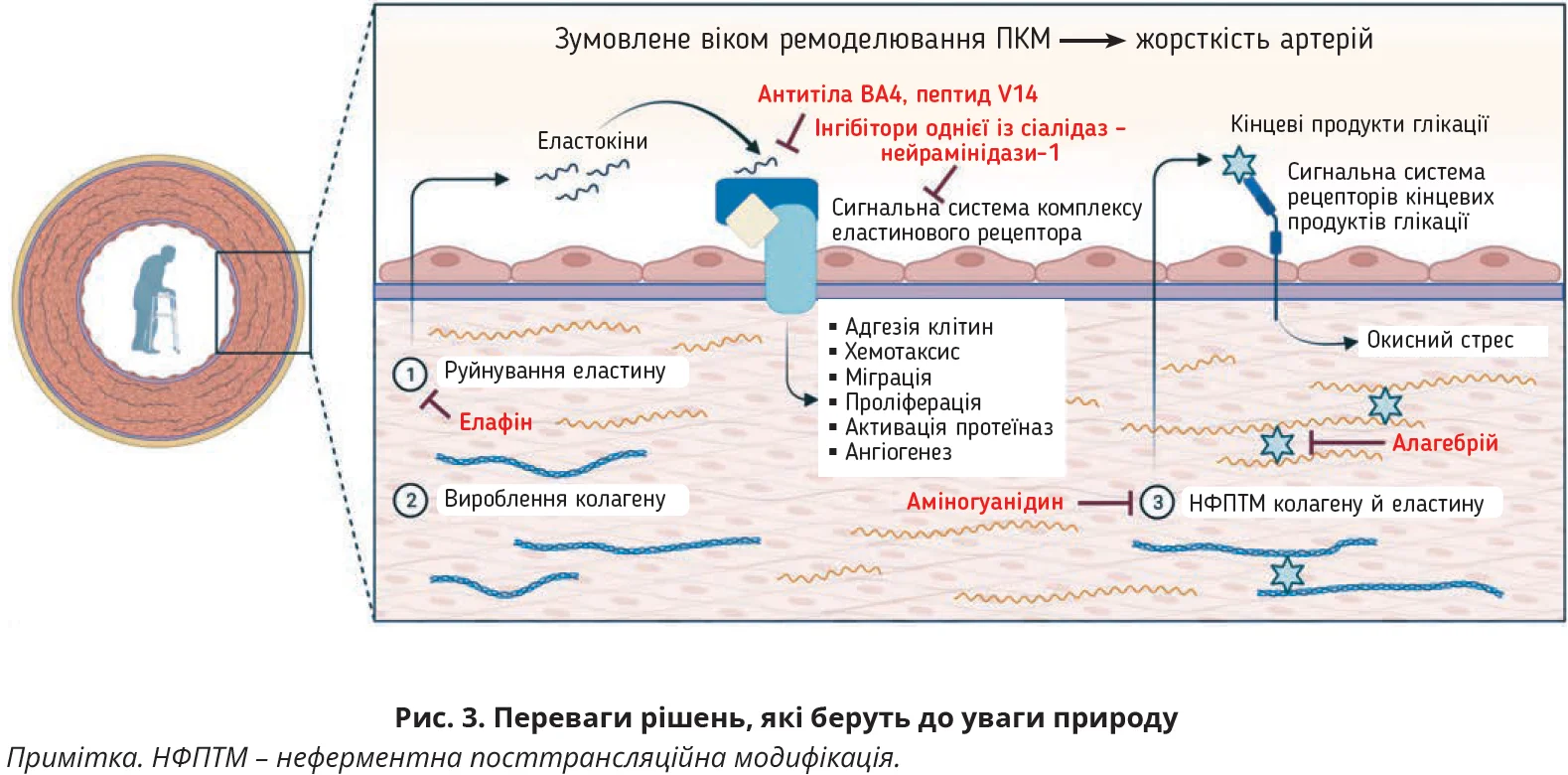
Білки ПКМ також підлягають таким хімічним реакціям, як неферментна посттрансляційна модифікація (НФПТМ, рис.), у ході якої метаболіти зв’язуються з відповідними функціональними групами (Jaisson and Gillery, 2010). Ці незворотні реакції мають кумулятивний характер і особливо стосуються білків із тривалим періодом життя (Gorisse et al., 2016). Найважливішими НФПТМ є глікація та карбамілювання. У ході глікації відбувається каскад окисних реакцій, який зумовлює утворення складних речовин – кінцевих продуктів глікації (Singh et al., 2001). НФПТМ спричиняє зміни структурних і функціональних властивостей білків ПКМ і незворотне накопичення продуктів посттрансляційної модифікації, що призводить до дезорганізації тканин судин (Goldin et al., 2006). Надалі продукти посттрансляційної модифікації можуть взаємодіяти з клітинними рецепторами, підсилюючи окисний стрес (Stirban et al., 2014). Продемонстровано, що і карбамілювання, і глікація беруть участь у старінні судин і розвитку довгострокових ускладнень хронічних захворювань, включаючи цукровий діабет і хронічну хворобу нирок (Machowska et al., 2016; Wang et al., 2007). Зокрема, карбамілювання еластичних волокон є молекулярним субстратом жорсткості аорти (Doue et al., 2021). У зв’язку з цим терапевтичний вплив на протеоліз еластинів і НФПТМ білків ПКМ може бути цінною стратегією відтермінування судинних ускладнень, асоційованих зі старінням.
Вплив на деградацію еластину за допомогою інгібіторів еластази залишається складним, оскільки еластин руйнує декілька різних протеїназ. Привабливою ціллю для терапевтичного впливу протягом тривалого часу вважаються матриксні металопротеїнази (ММП); станом на сьогодні розроблено та протестовано на тваринах цілу низку інгібіторів ММП. Однак потенціал для клінічного застосування продемонструвало лише кілька таких інгібіторів, передусім через широкий спектр субстратів дії та побічні ефекти (Vandenbroucke and Libert, 2014).
Мішенню фармакотерапії можуть також виступати катепсини, зокрема катепсин S (Figueiredo et al., 2015; Lai et al., 2020). Інгібітори катепсину S продемонстрували багатообіцяльні результати щодо збереження цілісності еластичних волокон і зменшення кількості судинних ускладнень, але ці препарати потребують подальшого вивчення в клінічних умовах. Іще однією стратегією є застосування ендогенних інгібіторів нейтрофільної еластази, наприклад елафіну (Alam et al., 2015; Zaidi et al., 2000).
Окрім запобігання руйнуванню еластичних волокон, може застосовуватися відновлення вже зруйнованих фібрил або неосинтез функціональних еластичних волокон (Kielty et al., 2002). Разючі результати було отримано для засобу, який відкриває АТФ-залежні K+-канали – міноксидилу. Застосування цього препарату в старих мишей протягом 3 місяців дало змогу зберегти цілісність еластичних пластинок у стінці судин і утворити нові еластичні волокна, що значно покращувало біомеханічні властивості артерій (Coquand-Gandit et al., 2017; Fhayli et al., 2019). Іншою багатообіцяльною можливістю є протидія руйнівним ефектам еластокінів за допомогою блокади їхньої взаємодії з ендоплазматичним ретикулумом. Цю функцію може виконувати або 14-мерний синтетичний пептид V14 (Blanchevoye et al., 2013), або блокувальні антитіла BA4 (Wrenn et al., 1986). Тривале призначення антитіл BA4 (протягом 2 місяців) мишам з експериментально змодельованим синдромом Марфана запобігало фрагментації еластину та макрофагальній інфільтрації аорти (Guo et al., 2013). Може застосовуватися й інгібування каталітичної активності білка NEU1 за допомогою інгібіторів сіалідази широкого спектра чи селективних інгібіторів цього білка. Серед останніх проводиться вивчення C9-BA-DANA та CG14601, які здатні достовірно відтерміновувати утворення жирових включень у корені аорти (Demina et al., 2021). Оскільки NEU1 утворює димери на плазматичній мембрані (Maurice et al., 2016), блокада його димеризації за допомогою специфічних пептидів зменшує каталітичну активність цього білка (Albrecht et al., 2020).
Більшість експериментів, присвячених протидії несприятливим ефектам НФПТМ, фокусуються на обмеженні утворення продуктів цього процесу. Основною стратегією є застосування конкурувальних засобів, як-от аміногуанідину – інгібітора глікації, котрий ефективно зменшує жорсткість артерій у щурів (за даними імпедансометрії та вазодилятаторної відповіді на ацетилхолін) (Chang et al., 2006; Li et al., 1996). Однак застосування цього препарату обмежене з міркувань безпеки (Bolton et al., 2004).
Своєю чергою, на процес карбамілювання можна вплинути за допомогою використання амінокислот, які зменшують карбамілювання білків у пацієнтів з уремією (Kalim et al., 2015). Крім того, розроблено сполуки, що руйнують перехресні зв’язки в кінцевих продуктах глікації, наприклад алагебрій. Незважаючи на певні позитивні результати, отримані в пацієнтів із серцево-судинними захворюваннями, алагебрій не став комерційно доступним як фармакопрепарат, насамперед у зв’язку з вартістю виробництва та труднощами ліцензування (Toprak and Yigitaslan, 2019), але й і через наявність небажаних побічних ефектів (Oudegeest-Sander et al., 2013).
Кальцифікація
Кальцифікація медії великих і середніх артерій, переважно у формі утворення кристалів гідроксіапатиту, є провідною ознакою поширених багатофакторних захворювань (наприклад, хронічної хвороби нирок, серцево-судинних захворювань) і старіння. У зв’язку з цим антикальцифікувальні засоби мають потенціал новітніх лікарських препаратів. Одним із потужних ендогенних інгібіторів судинної кальцифікації є неорганічний пірофосфат плазми, який утворюється з трифосфатів нуклеотидів, переважно АТФ, у ході синхронізованого функціонування трьох ферментів. Близько 60-70% АТФ, який міститься в циркуляції, вивільняється з печінки та транспортується АТФ-зв’язувальним касетним транспортером ABCC6 (Jansen et al., 2014). Згодом АТФ гідролізується до неорганічного пірофосфату й АМН за допомогою ендонуклеотидної пірофосфатази / фосфодіестерази-1 (Rutsch et al., 2003) – єдиного ферменту, здатного продукувати позаклітинний пірофосфат. Плазмовий неорганічний пірофосфат перетворюється на прокальцифікувальний неорганічний фосфат під впливом мембранозв’язаних і циркулювальних лужних фосфатаз, передусім тканинонеспецифічної лужної фосфатази та плацентарної лужної фосфатази (лише у вагітних) (Veiga-Lopez et al., 2020). АМФ далі розкладається на аденозин і неорганічний фосфат. Аденозин інгібує експресію тканинонеспецифічної лужної фосфатази, що робить його потенційним засобом впливу на кальцифікацію судин (Goettsch et al., 2022).
Кальцифікація медії судин також тісно пов’язана з ушкодженням ДНК внаслідок окисного стресу.
У таких умовах у стінці судин накопичується полі-АДФ-рибоза, яка спричиняє процес біомінералізації через концентрацію кальцію з утворенням ядер нуклеації (Müller et al., 2019).
Модель прогерії (прискореного старіння) на мишах виявила, що надмірна судинна кальцифікація асоціюється з підвищеним рівнем тканинонеспецифічних лужних фосфатаз і зниженням плазмового вмісту АТФ і неорганічного пірофосфату приблизно на 90% (Villa-Bellosta, 2019). Це додатково доводить зв’язок між старінням і порушенням балансу між неорганічним пірофосфатом і неорганічним фосфатом. Для подальшого розуміння механізмів кальцифікації судин і створення препаратів, здатних їй протидіяти, доцільно вивчати рідкісні генетичні захворювання (еластичну псевдоксантому; генералізовану кальцифікацію артерій дитячого віку).
Багатообіцяльним підходом до зменшення кальцифікації судин і поширеності асоційованих із нею вікових захворювань є відновлення вже згадуваного балансу між неорганічним пірофосфатом і неорганічним фосфатом. Із цією метою запропоновано застосовувати пероральні добавки неорганічного пірофосфату й інгібітор тканинонеспецифічної лужної фосфатази ланзопразол (ці препарати нині досліджуються в осіб з еластичною псевдоксантомою), а також розчинний рекомбінантний фермент INZ-701 (досліджується в осіб із генералізованою кальцифікацією артерій дитячого віку). Ефект неорганічного пірофосфату відтворює також міоінозитолгексафосфатаза (Sinha et al., 2022). При генералізованій кальцифікації артерій дитячого віку успішно застосовуються негідролізовані стабільні аналоги неорганічного пірофосфату – бісфосфонати, наприклад етидронат (Rutsch et al., 2008). Останній досліджується й за еластичної псевдоксантоми (Bartstra et al., 2020; Kranenburg et al., 2018).
На додаток до відновлення балансу неорганічних фосфату та пірофосфату потенційною терапевтичною мішенню може бути вивільнення АТФ, на яке можна впливати через АТФ-зв’язувальний касетний транспортер ABCC6. Фармакошаперон 4-фенілбутират, схвалений для усунення розладів циклу сечовини, здатен корегувати внутрішньоклітинну локалізацію неправильно розташованих мутантних білків-транспортерів (Le Saux et al., 2011). Пероральне застосування цієї речовини вже досліджено для низки рідкісних захворювань, асоційованих із дефектами АТФ-зв’язувальних транспортерів (Gonzales et al., 2015), і може стати методом лікування еластичної псевдоксантоми (Le Saux et al., 2011; Pomozi et al., 2017).
Крім наведених способів, опосередковану окисним стресом кальцифікацію можна зменшити за допомогою інгібування полі-АДФ-рибозної полімерази. Інгібітором останньої виступає, зокрема, міноциклін – напівсинтетичний тетрациклін другого покоління, котрий наразі застосовується при акне. Показано, що міноциклін ефективно протидіє ектопічній мінералізації в моделях еластичної псевдоксантоми в мишей і рибок даніо (Bouderlique et al., 2022; Huang et al., 2022; Nollet et al., 2022).
Загалом поточні дослідження фокусуються на кальцифікації судин у разі рідкісних генетичних захворювань. Для валідації таких стратегій за серцево-судинних захворювань та інших хвороб, пов’язаних із віком, потрібні подальші дослідження.
Література
Roth L., Dogan S., Tuna B.G., Aranyi T., Benitez S., et al. Pharmacological modulation of vascular ageing: a review from VascAgeNet. Ageing Res. Rev. 2023 Dec; 92: 102122. doi: 10.1016/j.arr.2023.102122. Epub 2023 Nov 11. PMID: 37956927.