Метаболічний синдром з ураженням серця, нирок, печінки та діабетом (CARDIAL-MS) – новий термін для інтеграції багатосистемної метаболічної хвороби
Переклала й адаптувала канд. мед. наук Ольга Королюк
Огляд присвячений опису відомих патофізіологічних взаємозв’язків метаболічного синдрому (МС), серцево-судинних захворювань (ССЗ), хронічної хвороби нирок (ХХН), метаболічно-асоційованої стеатотичної хвороби печінки (МАСХП), переддіабету та діабету 2-го типу (Д2Т), пропонуючи нову модель CARDIAL-MS (CArdio-Renal-DIAbetes-Liver-Metabolic Syndrome) – МС з ураженням серця, нирок, печінки й діабетом.
Кардіоренальні метаболічні хвороби (КРМХ) мають складну патофізіологію, що включає численні кардіометаболічні фактори ризику та впливи інших систем. Нещодавно Американська асоціації серця запровадила концепцію кардіоренального МС, який пов’язує ожиріння, Д2Т, МС з подальшим виникненням ССЗ і ХХН. У цьому контексті також розглядалася МАСХП, яка посилює інсулінорезистентність (ІР) і системне запалення. Проте печінка є метаболічним органом, що може визначати появу діабету та КРМХ.
Для розширення розуміння патофізіології ризику ураження органів-мішеней запропоновано нову концепцію CARDIAL-MS, яка охоплює: 1) адипопатію (надлишок або несприятливий розподіл жиру); 2) ектопічне відкладення ліпідів у печінці, м’язах, підшлунковій залозі, епікардіальній і периренальній жировій тканині (ЖТ); 3) вплив адипо-/гепатокінів. Розширена патофізіологічна модель CARDIAL-MS пояснює взаємодію ключових механізмів КРМХ – основної причини смерті у світі.
Адипопатія – пусковий механізм CARDIAL-MS
Ожиріння (індекс маси тіла (ІМТ) ≥30 кг/м²) і асоційовані стани досягли масштабів пандемії. За період 1980-2014 рр. поширеність ожиріння подвоїлася в 73 країнах. Станом на 2021 р. надмірну масу тіла (ІМТ – 25-29,9 кг/м²) або ожиріння зафіксовано в 1,00 мільярда чоловіків та 1,11 мільярда жінок. За прогнозами, до 2030 р. близько 50% дорослого населення планети житиме з надмірною масою тіла чи ожирінням.
Масштабний аналіз 12 систематичних оглядів, 53 метааналізів і 12 менделівських рандомізованих досліджень за участю 30 мільйонів осіб підтвердив причинно-наслідковий зв’язок між ожирінням і ССЗ. Установлено безпосередні зв’язки ІМТ з гіпертензією, ішемічною хворобою серця (ІХС), серцевою недостатністю (СН), фібриляцією передсердь (ФП), інсультом, стенозом аортального клапана, легеневою емболією та венозною тромбоемболією. Усі ускладнення, за винятком інсульту, мали причинно-наслідкові зв’язки з ожирінням.
Проте пов’язані з ожирінням метаболічні розлади не обов’язково присутні в людини з надлишком маси тіла. І навпаки, в осіб з нормальним ІМТ також можливий ризик ускладнень, асоційованих з ожирінням. Імовірним поясненням такого парадоксу є розподіл жиру в організмі та його ектопічне накопичення. Існує два жирові депо: підшкірна ЖТ (ПЖТ) і вісцеральна ЖТ (ВЖТ). Завдяки розширюваності та пластичності належним місцем депонування жиру є ПЖТ, а жир на сідницях і стегнах – глутеофеморальна ЖТ (ГФЖТ) сприятливо впливає на розлади метаболізму. ВЖТ не є належним місцем накопичення жиру й патофізіологічно пов’язана з ІР і МС. Широке впровадження магнітно-резонансної томографії (МРТ) та комп’ютерної томографії (КТ) дало змогу визнати ВЖТ незалежним фактором ризику Д2Т, ССЗ й асоційованої з ними смертності. Нещодавній аналіз результатів МРТ усього тіла 40 032 осіб підтвердив, що ВЖТ асоціюється з підвищеним ризиком Д2Т й ІХС; ГФЖТ асоціюється зі зниженням ризику; абдомінальна ПЖТ переважно нейтральна. Отже, першочергове значення для метаболічного здоров’я має ГФЖТ, дефіцит якої обмежує місткість депонування жиру, що етіологічно та генетично асоціюється з ІР і КРМХ.
Специфічні локуси поблизу або всередині L3MBTL3, DNAH10 і CCDC92 впливають на експресію генів ЖТ, що порушує адипогенез, зменшує периферичні жирові депо, посилюючи ризик КРМХ. Поряд з генетичною схильністю до несприятливого розподілу жиру певну роль відіграють зовнішні чинники, що призводять до ожиріння: широка доступність і низька вартість енергетично щільних продуктів та обмежена фізична активність унаслідок урбанізації. Коли здатність до розширення ПЖТ вичерпана, відкладання жиру в ній припиняється, що спричиняє вісцеральне й ектопічне відкладання ліпідів у тканинах, не придатних для накопичення жиру: печінці, м’язах, підшлунковій залозі, серці та нирках. Дисфункціональна, резистентна до інсуліну ЖТ зумовлює ектопічне відкладання ліпідів шляхом вивільнення вільних жирних кислот (ВЖК). Отже, ВЖТ можна вважати ектопічним жировим депо через збільшення доступності циркулювальних ВЖК, які спрямовуються до ектопічних ділянок (рис. 1).
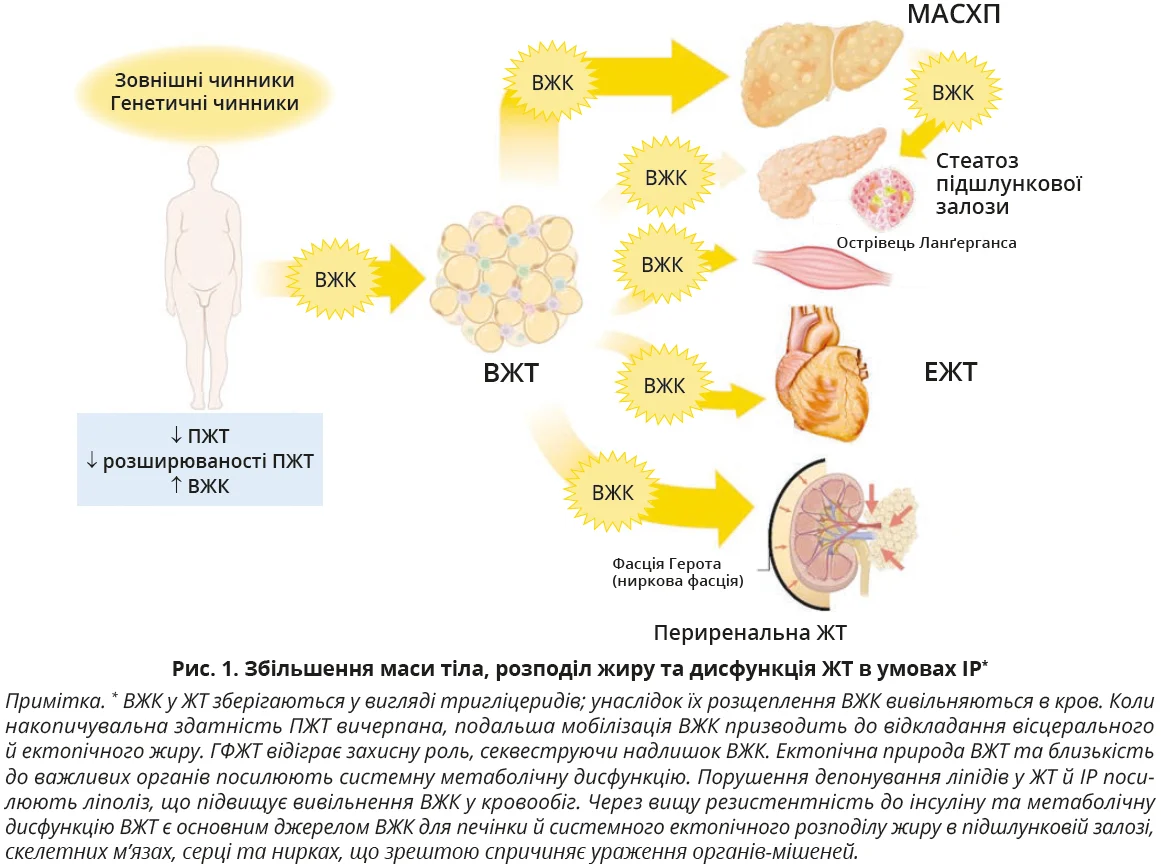
Нещодавнє дослідження із застосуванням клемп-тесту, МРТ черевної порожнини та біопсій абдомінальної ПЖТ і м’язів продемонструвало, що пригнічення опосередкованого інсуліном співвідношення жирних кислот прямо корелює з інсулінозалежним поглинанням глюкози, але негативно корелює з печінковим жиром і ВЖТ.
Ектопічне відкладання жиру запускає порушення метаболічного гомеостазу: сигналізацію адипо-/гепатокінів, запалення, посилення ІР, порушення секреції інсуліну, ендотеліальну дисфункцію, пошкодження тканин і фіброз, тобто патологічні процеси, що призводять до Д2Т, МАСХП і КРМХ.
Ектопічні жирові депо: порушення метаболічного гомеостазу при CARDIAL-MS
Стеатотична хвороба печінки
Жирова дистрофія, або стеатотична хвороба печінки, є найпоширенішою патологією печінки, яка вражає третину населення світу. Через часту асоціацію з ІР, ожирінням і Д2Т запропоновано терміни «МАСХП» і «МАСГ» (метаболічно-асоційований стеатогепатит). За визначенням, МАСХП асоціюється принаймні з одним з п’яти кардіометаболічних факторів ризику: 1) ІМТ ≥25 кг/м² чи окружність талії (ОТ) >90 см у чоловіків, >80 см у жінок або еквівалент за етнічною приналежністю; 2) переддіабет або Д2Т; 3) артеріальний тиск ≥130/85 мм рт. ст. або специфічна антигіпертензивна терапія; 4) тригліцериди плазми крові ≥150 мг/дл або ліпідознижувальна терапія; 5) холестерин ліпопротеїнів високої щільності ≤40 мг/дл у чоловіків, ≤50 мг/дл у жінок або ліпідознижувальна терапія. МАСХП вважається печінковим проявом МС.
З’являється дедалі більше доказів потенційного причинно-наслідкового зв’язку між MAСХП і ССЗ. Метааналіз 36 проспективних когортних досліджень за участю понад 5,8 мільйона осіб середнього віку показав, що МАСХП асоціюється з підвищеним ризиком летальних і нелетальних серцево-судинних ускладнень (відносний ризик (ВР) 1,45; 95% довірчий інтервал (ДІ) 1,31-1,61) незалежно від віку, статі, показників ожиріння, Д2Т й інших кардіометаболічних факторів ризику. Ризик ССЗ зростав у тяжчих випадках МАСХП з вищим ступенем фіброзу печінки (ВР 2,50; 95% ДІ 1,68-3,72). Причинами смерті пацієнтів з МАСХП є специфічні печінкові ускладнення та позапечінкові причини, зокрема ССЗ. Аналіз даних 13 099 пацієнтів з МАСХП і групи контролю показав вищу смертність від усіх причин у групі МАСХП (ВР 1,85; 95% ДІ 1,74-1,96); найвищі 15-річні кумулятивні частки смертності були зумовлені раком (окрім гепатоцелюлярної карциноми) та ССЗ – 7,3 та 7,2% відповідно.
Чітко причинно-наслідковий зв’язок між МАСХП і ССЗ можуть встановити лише менделівські рандомізовані дослідження, що використовують генетичні варіанти для оцінювання причинних ефектів. Дослідження асоціації на рівні всього геному ідентифікувало 94 незалежні локуси, асоційовані з МАСХП; полігенна модель оцінювання ризику підтвердила значущий причинний вплив МАСХП на ІХС. Інше подібне дослідження виявило стійкий зв’язок між генетично передбачуваним підвищенням сироваткових рівнів аланінамінотрансферази (АЛТ), МАСХП та ІХС.
Дослідження поширених генетичних варіантів, які асоціюються з МАСХП, переконливо свідчать, що ліпіди плазми крові по-різному впливають на ризик ССЗ. Асоціації однонуклеотидних поліморфізмів, які впливають на потік жирних кислот і ліпогенез de novo з ефектом підвищення ліпідів (наприклад, GCKR), посилюють ризик ІХС. У носіїв генетичних варіантів MTTP, PNPLA3, TM6SF2, що зумовлюють МАСХП через порушення секреції ліпопротеїнів дуже низької щільності (ЛПДНЩ), спостерігався більш кардіозахисний фенотип через нижчі рівні ліпідемії. Повногеномне дослідження виявило 13 генетичних варіантів, що асоціюються зі збільшенням умісту жиру в печінці. Варіанти, пов’язані з посиленим ліпогенезом de novo, показали, що алелі TRIB1, GCKR, ADH1B та CDHR4 асоціюються з вищим ризиком ІХС й інфаркту міокарда. Варіанти, пов’язані з порушенням печінкового експорту тригліцеридів (алелі PNPLA3, TMS6SF2, APOE, SUGP1) асоціюються з нижчим ризиком ІХС й інфаркту міокарда, проте вищим ризиком Д2Т.
Отже, зв’язок між МАСХП і ССЗ не лише епідеміологічний, а й причинно-наслідковий, незважаючи на генетичну гетерогенність. Генетично підвищений уміст жиру в печінці посилює ризик Д2Т, цирозу, гепатоцелюлярної карциноми, раку внутрішньопечінкових жовчних проток і жовчного міхура, демонструючи дозозалежний зв’язок незалежно від механізму.
МАСХП як причина ССЗ: посередницькі чинники
Підвищений приплив ВЖК до печінки є наслідком адипопатії й ІР, тобто підвищеного вивільнення ВЖК та зниження їх поглинання скелетними м’язами.
Це, своєю чергою, погіршує дисрегуляцію передачі сигналів інсуліну, утворюючи замкнене коло, що стимулює печінковий ліпогенез de novo та посилює накопичення ліпідів у печінці. Тривалий вплив високих рівнів ВЖК і ліпогенезу de novo призводить до накопичення в печінці ліпотоксичних форм ліпідів, що спричиняють мітохондріальну дисфункцію, порушення автофагії, оксидативний стрес, активацію інфламасом, пошкодження гепатоцитів і апоптоз. Подальші репаративні реакції з активацією зірчастих клітин зумовлюють відкладання колагену, прогресування фіброзу та, зрештою, печінкову недостатність.
Тим часом МАСХП і ВЖТ спричиняють атерогенну дисліпідемію та гіпертригліцеридемію через: 1) підвищення печінкової секреції великих, збагачених тригліцеридами молекул ЛПДНЩ; 2) порушення кліренсу збагачених тригліцеридами ліпопротеїнів, підвищення рівнів аполіпопротеїну С-III (АпоС-III) з тривалим перебуванням у кровообігу до гідролізу ліпопротеїнліпазою (рис. 2). Тривале перебування в судинах підвищує ймовірність проникнення ліпопротеїнів у ендотелій, утворення атеросклеротичних бляшок і місцевого запалення. Спричиняючи атерогенну дисліпідемію та субклінічне запалення, МАСХП і ектопічна адипопатія посилюють ризик нестабільності атеросклеротичних бляшок.
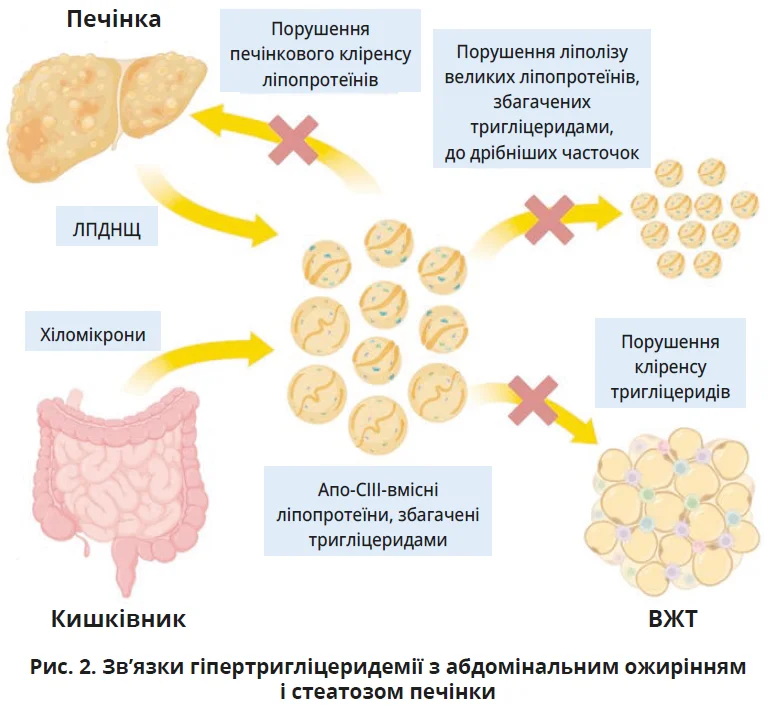
Добре відомо, що в умовах ожиріння ВЖТ індукує запалення. Імовірно, запалення спричиняє гепатокін – циркулювальний фактор печінкового походження. Клінічно очевидну взаємодію між печінкою та ВЖТ за метаболічних розладів підтверджено на тваринній моделі: ожиріння в мишей стимулювало гепатоцити до синтезу та секреції дипептидилпептидази-4 (ДПП-4), яка взаємодіяла з плазмовим фактором Xa, зумовлюючи ІР і запалення. Розчинна ДПП-4 активувала шлях кавеоліну-1 у макрофагах ЖТ. Крім того, фактор Xa активував шлях активованого протеазою рецептора-2. Обидва шляхи синергічно стимулювали позаклітинні сигнально-регульовані протеїнкінази 1 і 2, а також ядерний фактор κB – дистальні сигнальні молекули запалення. Придушення експресії ДПП-4 у гепатоцитах пригнічувало запалення ВЖТ й ІР, тоді як фармакологічне пригнічення ДПП-4 не демонструвало такого ефекту. Отже, через гепатокіни печінка може ініціювати ІР і запалення ЖТ, що потенційно призводить до хронічних захворювань, асоційованих з ожирінням.
Вісь кишківник – печінка – ЖТ при CARDIAL-MS
Вісь кишківник – печінка – ЖТ пов’язує мікробіоту людини з MAСХП і адипопатією. Кишкова мікробіота є «невидимим органом», взаємопов’язаним з різними тканинами. Печінка є основним органом, що забезпечує відтік крові з кишківника, підтримуючи постійний зв’язок зі станом мікробіоти. Крім того, печінка з’єднується з кишківником через жовчні шляхи.
Загальновідомо, що продукти життєдіяльності кишківника, зокрема метаболіти макро- та/або мікроорганізмів, а також пов’язані з мікробами молекулярні структури, транспортуються до печінки, модулюючи її функції. Існують докази складної взаємодії між кишковою мікробіотою, кишківником і ЖТ, що впливає на інсулін. Комплексне дослідження, котре вивчало транскриптоміку кишківника, печінки та різних видів ЖТ, виявило негативну кореляцію між кількома видами Proteobacteria phylum і чутливістю до інсуліну; транскриптомічний аналіз порожньої, клубової та товстої кишок виявив Т-клітинні сигнатури, прямо пов’язані з чутливістю до інсуліну. Багаторівнева оміксна та тканинна інтеграція пов’язує Proteobacteria з дезоксихолевою кислотою порожньої кишки, а також з генами порожньої кишки та ВЖТ, що беруть участь у сигнальних шляхах актинового цитоскелета, інсуліну й Т-клітин. Установлено, що рівень глікемії натще стабільно асоціювався з генами, індукованими інтерфероном, і противірусними відповідями у ВЖТ та кишківнику. Нещодавно виявлено часте підвищення вмісту уробіліну в сечі в осіб із ССЗ, що вказує на взаємодії між жовчними кислотами сироватки крові та ССЗ.
Отже, печінка може відігравати основну роль у патофізіології CARDIAL-MS, що пояснюється складними взаємозв’язками між мікробіотою, MAСХП і адипопатією.
Чи може печінка бути причиною Д2Т?
Поширеність МАСХП серед людей з Д2Т в середньому сягає 65,3% (від 53,1% в Африці до 80,6% у Східній Європі).
Обсерваційні дослідження показали, що помірне підвищення рівнів трансаміназ і g-глутамілтрансферази (ГГТ) асоціюється з вищим ризиком Д2Т у майбутньому. Зв’язок між МАСХП і частотою виникнення Д2Т підтверджено результатами кількох метааналізів. Зокрема, метааналіз, що включав 20 досліджень за участю 117 020 пацієнтів, використовував АЛТ, аспартатамінотрансферазу (АСТ), ГГТ та наявність ультрасонографічних ознак стеатозу печінки як діагностичні критерії МАСХП; відносні ризики Д2Т для кожного зі вказаних критеріїв становили, відповідно, 1,97 (95% ДІ 1,80-2,15), 1,58 (95% ДІ 1,43-1,74), 1,86 (95% ДІ 1,71-2,03) та 1,86 (95% ДІ 1,76-1,95). Інший метааналіз, що включав 33 дослідження за участю 501 022 осіб, продемонстрував вищий ризик Д2Т серед пацієнтів з МАСХП порівняно з особами без МАСХП (2,19; 95% ДІ 1,93-2,48); у підгрупі зі значним ступенем фіброзу печінки ризик був іще вищим (3,42; 95% ДІ 2,29-5,11). Нарешті, комплексний систематичний аналіз 129 досліджень за участю тисяч учасників виявив істотно вищі ризики виникнення Д2Т (2,56; 95% ДІ 2,10-3,13), переддіабету (1,69; 95% ДІ 1,22-2,35) і МС (2,57; 95% ДІ 1,13-5,85) у групі MAСХП порівняно
з групою без MAСХП; найвищий ризик Д2Т виявлено в підгрупі пацієнтів з тяжчим ураженням печінки (3,60; 95% ДІ 2,10-6,18). Отже, між частотою виникнення Д2Т та наявністю й тяжкістю МАСХП існує зв’язок «доза – відповідь».
І навпаки, Д2Т обтяжує перебіг MAСХП. У дослідженні за участю 713 пацієнтів з МАСХП підвищення HbA1c на кожен 1% збільшувало ймовірність вищої стадії фіброзу печінки через 1 рік на 15%. Окрім того, Д2Т підвищує ризик ускладнень, пов’язаних із цирозом печінки. У ретроспективному дослідженні наявність Д2Т у пацієнтів з MAСХП асоціювалася з високим ризиком гепатоцелюлярної карциноми (3,21; 95% ДІ 1,09-9,50).
Незважаючи на чіткі патофізіологічні й епідеміологічні зв’язки між MAСХП і Д2Т, висновок щодо існування причинно-наслідкового зв’язку важко зробити, оскільки більшість досліджень були обсерваційними. Для визначення ймовірного причинно-наслідкового ефекту біомаркерів функції печінки або печінкового жиру деякі дослідники застосували менделівську рандомізацію. Зокрема, аналіз даних 64 094 пацієнтів з Д2Т та 607 012 осіб групи контролю з використанням генетичних варіантів, що асоціюються з печінковими ферментами та Д2Т або інсуліном натще, встановив зв’язок між генетично передбаченим підвищенням рівнів циркулювальних трансаміназ і вищим ризиком Д2Т. Генетична схильність до гіперінсулінемії натще корелювала з підвищенням АЛТ. Отримані результати вказують на MAСХП як причину Д2Т в умовах ІР.
Ба більше, ці висновки підтверджують гіпотезу подвійного циклу (див. нижче).
Аналізи чутливості з використанням генетичних варіантів, пов’язаних з об’ємом печінки та підшлункової залози, а також умістом жиру за даними МРТ продемонстрували, що печінковий жир і менший об’єм підшлункової залози є причинними факторами підвищеного ризику виникнення Д2Т. Натомість більший об’єм підшлункової залози асоціювався зі зниженням ризику на 24% на кожне стандартне відхилення; генетичного зв’язку між стеатозом підшлункової залози та ризиком виникнення Д2Т не виявлено.
Гепатокіни: білки печінкового походження, пов’язані з КРМХ
Печінка виробляє та вивільняє приблизно 13 білків, пов’язаних з метаболічними розладами. Принаймні 6 із них можуть бути особливо значущими для CARDIAL-MS: фетуїн A, фактор росту фібробластів-21 (FGF-21), глобулін, що зв’язує статеві гормони (SHBG), ангіопоетиноподібний білок-3, селенопротеїн P та ДПП-4. Серед них лише SHBG та FGF-21 знижені при стеатотичній хворобі печінки. Більшість гепатокінів асоціюються з ІР, запаленням, дисфункцією β-клітин і кардіометаболічним ризиком незалежно від ожиріння.
Асоціацію з виникненням Д2Т доведено для фетуїну A – гепатокіну, який переважно синтезується печінкою та відіграє вирішальну роль у багатьох важливих процесах, як-от передача сигналів рецепторів інсуліну, дисфункція адипоцитів, запалення, фіброз печінки, ліпідна токсичність, вироблення тріацилгліцеролів, модифікація фенотипу макрофагів, стимуляція ангіогенезу, пошкодження β-клітин, апоптоз і активація Toll-подібного рецептора-4. Рівень фетуїну А підвищений у разі MAСХП, асоціюється з МС і його компонентами. Цікаво, що підвищення рівня фетуїну А передує початку МС. Декілька обсерваційних досліджень установили кореляцію між фетуїном А та ризиком виникнення Д2Т.
Утім, результати досліджень із застосуванням менделівської рандомізації залишаються суперечливими. Аналіз даних дослідження EPIC-InterAct за участю 12 403 пацієнтів з нещодавно встановленим Д2Т не виявив сильного зв’язку між генетичними показниками однонуклеотидних поліморфізмів у гені AHSG, що кодує фетуїн А, та випадками Д2Т. Проте дослідження з більшою кількістю учасників, тривалішим періодом спостереження та використанням нових генетичних варіантів, пов’язаних з фетуїном А, виявило, що в осіб з генетично зумовленим підвищенням рівня фетуїну A ризик виникнення Д2Т є на 21% вищим.
Оскільки пацієнти з MAСХП переважно вмирають від ССЗ, досліджувалися потенційні зв’язки між ССЗ та фетуїном А. Автори аналізу 12 обсерваційних досліджень дійшли висновку, що цей гепатокін є ймовірним медіатором, оскільки 7 досліджень повідомляли про його зв’язок з різними серцево-судинними проявами.
Стеатоз підшлункової залози
Стеатоз підшлункової залози – це загальний термін для опису накопичення жиру в залозі від жирової інфільтрації до запалення та фіброзу. Найчастіше спостерігається інфільтрація залози адипоцитами, іноді – внутрішньоклітинне накопичення крапель жиру. Жирова інфільтрація може бути зворотною, а також незворотною внаслідок загибелі ацинарних клітин (так зване ліпозаміщення підшлункової залози).
Уперше стеатоз підшлункової залози описано в 1933 р. У 1980-х дані автопсії показали, що заміщення жиром понад 25% залози асоціюється з вищим ризиком генералізованого атеросклерозу та Д2Т. За даними 11 досліджень за участю 12 675 осіб, поширеність стеатозу підшлункової залози становить 33%. До накопичення жиру в залозі призводять збільшення маси тіла, переддіабет або Д2Т, МС, МАСХП, похилий вік і вживання алкоголю. Наявність ≥1 з компонентів МС асоціювалося зі збільшенням поширеності стеатозу підшлункової залози на 37%. Інші можливі чинники охоплюють чоловічу стать, низьку масу тіла при народженні та моногенні хвороби, зокрема діабет 8-го типу в зрілому віці (MODY8), пов’язаний з тяжким панкреатичним ліпоматозом з дитинства.
Найчастіше адипоцити інфільтрують екзокринну частину залози. Проте можлива ліпідна інфільтрація острівців Ланґерганса, особливо в осіб старшого віку, в разі Д2Т або часткової сімейної ліподистрофії 2-го типу. Рой Тейлор запропонував гіпотезу подвійного циклу, що пов’язує стеатоз печінки та підшлункової залози з патогенезом Д2Т. Гіперпродукція тригліцеридів печінкою призводить до накопичення жиру в підшлунковій залозі; внаслідок ліпотоксичності порушується функція β-клітин, знижується секреція інсуліну та виникає гіперглікемія. Пізніше гіпотезу Тейлора перевірено в серії досліджень, які продемонстрували відновлення функції β-клітин після обмеження калорій у дієті, що супроводжувалося швидким зниженням жиру в печінці та повільнішим зменшенням стеатозу підшлункової залози. Іншим шляхом відкладання жирних кислот у підшлунковій залозі, не залежним від вироблення тригліцеридів печінкою, може бути прямий потік ВЖК із ЖТ.
У систематичному огляді з метааналізом і метарегресією виявлено, що стеатоз підшлункової залози асоціюється з істотним підвищенням ризику Д2Т (2,08; 95% ДІ 1,44-3,00) та МС (2,37; 95% ДІ 2,07-2,71). Інше дослідження виявило, що худі люди зі стеатозом підшлункової залози мають більшу ймовірність виникнення Д2Т впродовж 6 років, аніж особи без стеатозу, чого не спостерігалося в людей з ожирінням. Отже, різні фенотипи Д2Т по-різному асоціюються з панкреатичним стеатозом.
Цікаво, що стеатоз підшлункової залози як такий не порушує секрецію інсуліну. В експерименті адипоцити підшлункової залози модулювали секрецію інсуліну, вивільняючи ВЖК; посилення секреції інсуліну відбувалося через рецептор-1 ВЖК у β-клітинах. Утім, за певних метаболічних обставин, як-от переддіабет, панкреатичний стеатоз асоціювався з порушенням секреції інсуліну. Очевидно, за таку взаємодію відповідають генетичні варіанти, що опосередковують ІР, а не секреторну здатність β-клітин. Отже, стеатоз підшлункової залози порушує секрецію інсуліну лише в осіб, генетично схильних до Д2Т. Імовірним механізмом є паракринна дія метаболітів або речовин, що секретуються панкреатичними адипоцитами, – цитокінів, хемокінів, адипокінів. Окрім того, тривалий вплив довголанцюгових насичених жирних кислот на β-клітини зумовлює внутрішньоклітинні зміни (утворення керамідів, оксидативний стрес, стрес ендоплазматичного ретикулуму, мітохондріальну дисфункцію, активацію протеїнкіназ), які погіршують секрецію інсуліну та гомеостаз глюкози, спричиняючи виникнення Д2Т.
Серцево-судинний стеатоз
Гіпертрофовані та збагачені тригліцеридами адипоцити зазнають клітинного стресу, що спричиняє ІР і запалення в ЖТ. Водночас активація Toll-подібних рецепторів і секреція прозапальних цитокінів (фактора некрозу пухлини-a – ФНП-a; інтерлейкіну-6 – ІЛ-6) надалі погіршують чутливість до інсуліну. Відтак посилюються ліполіз, вивільнення ВЖК у системний кровообіг та ектопічне накопичення ліпідів, що погіршує локальну ІР і запалення. Крім того, асоційовані з ІР і гіперінсулінемією порушення внутрішньоклітинного метаболізму посилюють накопичення жирних кислот усередині клітин, створюючи прозапальне середовище.
Ключовими мішенями для перенесення ВЖК у серцево-судинній системі є ендотелій, кардіоміоцити й епікардіальна ЖТ (ЕЖТ). Клітини ендотелію поглинають циркулювальні ВЖК, хіломікрони та ЛПДНЩ через альбумін і Toll-подібні рецептори; ліпопротеїнліпаза сприяє вивільненню ВЖК з ліпопротеїнів. Усередині ендотеліоцитів ВЖК перетворюються на тригліцериди під дією діацилгліцерол-О-ацилтрансферази. Клітини ендотелію здатні формувати й метаболізувати внутрішньоклітинні ліпідні краплі, які служать буферами проти ліпотоксичності та резервуарами ВЖК для власних потреб і сусідніх паренхіматозних клітин.
Мобілізацію ВЖК з тригліцеридів усередині ліпідних крапель ініціює жирова тригліцеридліпаза; порушення регуляції або дефіцит цього ферменту, а також надмірне споживання ліпідів призводять до накопичення ліпідних крапель, що спричиняє ендотеліальну дисфункцію через зниження експресії ендотеліальної синтази оксиду азоту, посилює стрес ендоплазматичного ретикулуму та запалення ендотелію. Усі вказані зміни зумовлюють прогресування атеросклерозу та підвищують судинний тонус, що призводить до артеріальної гіпертензії. У серійному дослідженні реципієнтів людського трансплантата серця внутрішньоклітинне накопичення тріацилгліцеролу та кераміду в кардіоміоцитах обернено корелювало з індексом HOMA-IR і асоціювалося з діастолічною й систолічною дисфункцією через 12 місяців після трансплантації незалежно від ІМТ.
Накопичення ліпідних крапель у міокарді відображає порушення гомеостазу жирних кислот і ліпотоксичність. У разі СН зі збереженою фракцією викиду (СНзбФВ) накопичення ліпідних крапель було вираженішим в осіб з ожирінням, аніж у пацієнтів з гіпертензією. Отже, за метаболічної патології (ожиріння, діабет) ліпотоксичність відіграє значнішу роль, аніж гіпертрофія й перевантаження тиском. Ожиріння тісно пов’язане з низкою ультраструктурних аномалій, як-от фіброз, деструкція саркомерів і мітохондрій, гіперактивність лізосом/фагосом.
ЕЖТ – це унікальне жирове депо між міокардом і вісцеральним епікардом, що переважно складається з адипоцитів, але містить запальні клітини (макрофаги, мастоцити), а також нервові, стромальні, судинні й імунні клітини. Загалом ЕЖТ класифікується як біла ЖТ, але має певні властивості бурої та бежевої ЖТ з експресією унікального транскриптому, відмінного від ВЖТ і ПЖТ. Крім того, ЕЖТ навколо лівого передсердя відрізняється від ЕЖТ, що інфільтрує коронарні артерії, по-різному впливаючи на прилеглі структури серця. Відсутність фасцій між ЕЖТ та міокардом зумовлює спільну мікроциркуляцію.
Роль ЕЖТ подвійна: 1) захисна термогенна функція полягає в забезпеченні серця теплом та енергією, максимально виражена в дітей; 2) негативна атерогенна й аритмогенна дія через пара- та вазокринну секрецію прозапальних і профібротичних цитокінів, притаманна старінню, ІХС, СН, ФП та Д2Т. Зокрема, при ІХС інфільтрована запальними клітинами коронарна ЕЖТ секретує цитокіни (моноцитарний хемоатрактантний білок-1, IЛ-6, ФНП-α) й адипокіни (хемерин, інтелектин-1, оментин-1, резистин, сергліцин) у просвіт коронарної артерії, посилюючи системне та локальне запалення в атеросклеротичних бляшках. Окрім того, ЕЖТ спричиняє виникнення ФП та її рецидиви після абляції. Збільшення об’єму або товщини ЕЖТ асоціювалося з порушеннями передсердної провідності. Імовірно, такі взаємодії охоплюють генетичні та нейронні фактори, запалення, фіброз, жирову інфільтрацію, структурне й електричне ремоделювання передсердь. Важливо, що асоціація між ЕЖТ та ФП частково або цілком не залежить від ожиріння. У патогенезі СН важливу роль відіграють механічні впливи фіброзно-жирових структур та ефекти ЕЖТ на запальні процеси, фіброз і вегетативну дисрегуляцію. Зв’язок між товщиною або об’ємом ЕЖТ і СНзбФВ суперечливий: повідомляється як про збільшення, так і про зменшення об’єму ЕЖТ порівняно зі здоровими особами. Тому поки що оцінювання об’єму ЕЖТ не має особливої клінічної користі.
Периренальна ЖТ, жир ниркового синуса та ХХН
Як відомо, ожиріння, МС, МАСХП, Д2Т та ССЗ пов’язані з різноманітними нирковими розладами. Добре відомими є епідеміологія та патофізіологія класичної діабетичної нефропатії. Активно досліджуються причинно-наслідкові зв’язки, що пов’язують ожиріння, МАСХП і МС з пошкодженням нирок. Нещодавні масштабні дослідження продемонстрували стійкий зв’язок між ожирінням і ХХН незалежно від діабету та гіпертензії. У когорті 1 405 016 дорослих ризик виникнення прогресивної ХХН логарифмічно зростав зі збільшенням ІМТ (надмірна маса тіла ® ожиріння ® морбідне ожиріння) незалежно від наявності Д2Т, гіпертензії або ССЗ. В іншому дослідженні за участю 337 422 європейців продемонстровано значущий причинно-наслідковий вплив генетичних ознак, пов’язаних з ІМТ, ОТ й ожирінням, на індекс здоров’я нирок – комплексний показник функції нирок. Прикметно, що артеріальний тиск і Д2Т становили лише 26-34% причинно-наслідкового зв’язку між ІМТ або ОТ й індексом здоров’я нирок. Відповідно, вплив ожиріння на здоров’я нирок залежить не тільки від гіпертензії та діабету. Інше масштабне дослідження з менделівською рандомізацією виявило сильний зв’язок між ожирінням і дисфункцією нирок, але значущого зв’язку з альбумінурією за відсутності Д2Т не було. Отже, існують різні та складні шляхи метаболічно-опосередкованого ураження нирок.
Кілька масштабних метааналізів підтвердили епідеміологічний зв’язок між MAСХП і ХХН. Ключове питання полягає в тому, чи є цей зв’язок наслідком спільних метаболічних і запальних шляхів, чи існує прямий патофізіологічний зв’язок, що спричиняє серцево-судинні ускладнення. Фактично ХХН значно підвищує ризик ССЗ через багатофакторний патофізіологічний процес, що включає гемодинамічні, нейрогормональні, метаболічні та запальні шляхи. Традиційні фактори ризику (гіпертензія, діабет, дисліпідемія, ожиріння) дуже поширені при ХХН і прискорюють атеросклероз. Активація ренін-ангіотензин-альдостеронової та симпатичної нервової систем спричиняє гіпертрофію лівого шлуночка й СН. Метаболічні порушення (накопичення уремічного токсину, кальцієво-фосфатний дисбаланс) посилюють кальцифікацію судин і фіброз міокарда. Крім того, ХХН індукує ендотеліальну дисфункцію та хронічне запалення, створюючи протромботичне та проатерогенне середовище. Дисфункція коронарних мікросудин і порушення біодоступності оксиду азоту спричиняють ішемію міокарда навіть за відсутності епікардіальної ІХС. Нарешті, в дослідженні EQUAL у двох незалежних когортах літніх людей із прогресивною ХХН ідентифіковано три білки (рецепторний білок-тирозинфосфатаза-σ, FCN2 й IGFBP6), пов’язані зі зниженням розрахованої швидкості клубочкової фільтрації (рШКФ). Отже, існує потенційний зв’язок між підвищеною експресією профібротичних білків, каскадом комплементу та прогресивною втратою функції нирок. У сукупності вказані механізми пояснюють значну поширеність ССЗ при ХХН. Проте встановити причинно-наслідковий зв’язок, ґрунтуючись на цих спостереженнях, складно через перекривання традиційних факторів ризику МАСХП і ХХН.
Гіпотеза щодо прямого причинно-наслідкового зв’язку між жировою інфільтрацією печінки та ХХН є відносно новою; накопичення доказів підтверджує її правдоподібність. Імовірними чинниками є генетична схильність, дисбактеріоз кишківника, печінкова ІР, низькоінтенсивне запалення, протромботичні стани, гіперурикемія, недостатнє вироблення адипонектину, активація ренін-ангіотензинової системи, порушення антиоксидантного захисту, нефротоксичність утворених печінкою метаболітів і секреція шкідливих гепатокінів.
Цікаво, що локальне накопичення ЖТ асоціюється з пошкодженням нирок і підвищеним ризиком МАСХП. Прикладами скупчення жиру навколо нирок є периренальна ЖТ та жир ниркового синуса. Периренальна ЖТ оточена щільною фіброзною мембраною; її надмір у малорозтяжному місці може спричинити інкапсуляцію та стискання нирки – гіпотеза ниркової тампонади. Ниркова капсула – серозний шар, що складається з колагену й еластину, охоплює більшу частину паренхіми нирок, перериваючись у воротах, де розміщені ниркова миска, нерви та судини. У воротах нирки периренальна ЖТ поширюється всередину, утворюючи жир ниркового синуса, який оточує вказані структури та дистальні частини сосочків. Жир ниркового синуса є унікальним різновидом ВЖТ й особливим типом периваскулярної ЖТ, що має васкуляризацію, іннервацію, лімфатичний дренаж і функціональну адренергічну систему. Надлишок жиру ниркового синуса може інфільтрувати та стискати ниркові сосочки й основні судини в ділянці воріт, пошкоджуючи нирку механічно або через ендокринні/паракринні впливи. Механічне стискання підвищує внутрішньонирковий тиск, погіршує ниркову перфузію, активує ренін-ангіотензинову систему, посилює реабсорбцію натрію, погіршує клубочкову фільтрацію та підвищує ризик ішемічного пошкодження. Крім того, жир ниркового синуса є метаболічно активним і реагує на системні сигнали через пряму ліпотоксичність, місцеві та системні адипо-/гепатокіни, оксидативний стрес і ендотеліальну дисфункцію, додатково посилюючи пошкодження нирок.
Нещодавно встановлено: запалення, спричинене периренальною ЖТ, є новим незалежним кардіометаболічним фактором ризику, що принаймні частково пояснює високий ризик серцево-судинних ускладнень на ранніх стадіях ХХН. Периренальна ЖТ містить суміш бурих і білих адипоцитів, які зазвичай проявляють імунорегуляторний фенотип у відповідь на запалення, але можливий перехід до запального фенотипу з гіперсекрецією прозапальних цитокінів і зниженим виробленням захисних адипокінів, зокрема адипонектину. Запальний зсув призводить до ендотеліальної дисфункції, фіброзу та пошкодження канальців, пов’язуючи дисфункцію периренальної ЖТ з прогресуванням ХХН.
Рисунок 3 деталізує складну патофізіологію CARDIAL-MS, яка охоплює адипопатію, ектопічне накопичення ліпідів і порушення регуляції сигналізації адипо-/гепатокінів, що зумовлюють Д2Т, МАСХП і КРМХ.
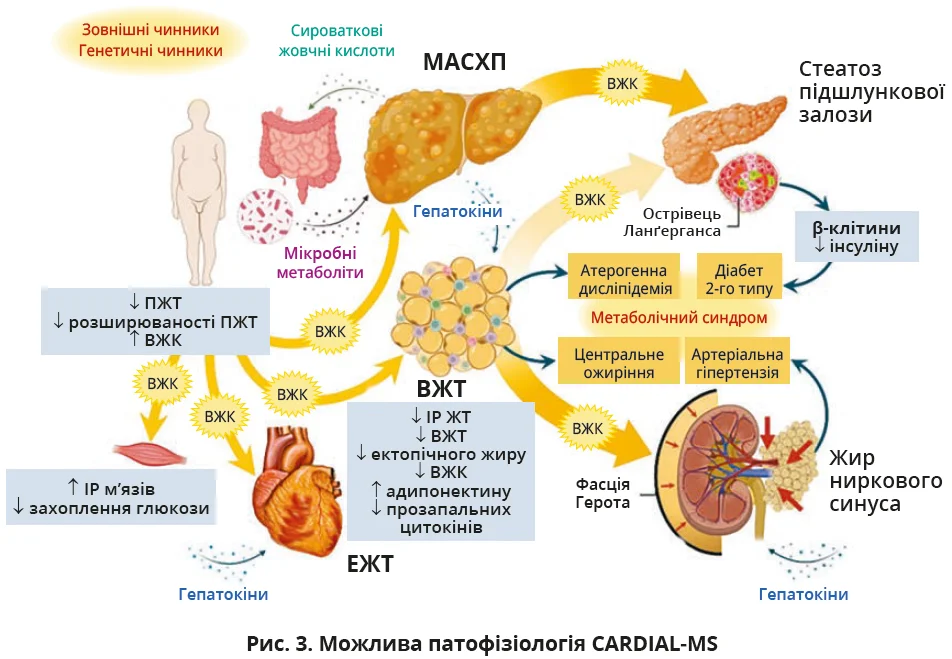
Фенотипи діабету, що дебютує в дорослому віці, й ектопічний жир
Окрім класифікації діабету на два типи, деякі дослідники пропонують нову концепцію субфенотипування діабету, що дебютує в дорослому віці. Субфенотипи з вищою ІР асоціюються з вищим ризиком розвитку ХХН. У дослідженні TUEF/TULIP визначено шість кластерів високого ризику Д2Т. Серед них 5-й кластер характеризувався надлишком ВЖТ та МАСХП, а 6-й – надлишком ВЖТ та жиру ниркового синуса; обидва кластери асоціювалися з вищим ризиком ХХН і смерті від ХХН. Отже, існують взаємозв’язки між Д2Т й ектопічним відкладанням жиру в печінці та навколо нирок.
Стадії CARDIAL-MS і методи візуалізації ектопічного жиру
Запропоновано чотири стадії CARDIAL-MS (рис. 4). Виявлення пацієнтів з високим ризиком метаболічних ускладнень може спростити ранню діагностику. Усім пацієнтам треба проводити антропометрію, оцінювати кардіометаболічні фактори ризику за критеріями МС Міжнародної діабетичної федерації (IDF), визначати глікемію натще та/або після навантаження для діагностики переддіабету та Д2Т.

Для візуалізації жиру в печінці й підшлунковій залозі найчастіше використовуються ультразвукове дослідження, КТ та МРТ. Для виявлення паранефрального жиру найінформативнішою є МРТ. На жаль, клінічні біомаркери ХХН (рШКФ, альбумінурія) інформативні на пізніх стадіях. Високу ефективність оцінювання стану нирок демонструють нові методи функціональної МРТ, даючи змогу оцінити об’єм нирок in vivo, швидкість клубочкової перфузії та склерозу, функцію, метаболізм, перфузію, оксигенацію, а також виявляти мікроструктурні зміни й фіброз на ранніх стадіях без уведення контрасту.
Висновки
Стадійна модель CARDIAL-MS класифікує пацієнтів у критичних точках переходу від оборотних метаболічних розладів до незворотного пошкодження органів і наголошує на важливості ранніх утручань для сповільнення прогресування КРМХ шляхом стратифікації ризику, ранньої діагностики та персоналізованих стратегій профілактики.
Література
Godoy-Matos A.F., Valério C.M., Júnior W.S.S., de Araujo-Neto J.M., Sposito A.C., Suassuna J.H.R. CARDIAL-MS (CArdio-Renal-DIAbetes-Liver-Metabolic Syndrome): a new proposition for an integrated multisystem metabolic disease. Diabetol. Metab. Syndr. 2025 Jun 16; 17 (1): 218. doi: 10.1186/s13098-025-01796-4.