Рекомендації Європейської тиреоїдної асоціації щодо діагностики й лікування генетичних розладів транспорту, метаболізму та дії тиреоїдних гормонів (2024). Частина 2
Переклала й адаптувала д-р мед. наук Лариса Стрільчук
Дефіцит монокарбоксилатного транспортера-8 (МКТ-8)
Вступ
Дефіцит МКТ-8 (синдром Аллана – Герндона – Дадлі) характеризується затримкою неврологічного розвитку різного ступеня, спричиненою мозковим гіпотиреозом, а також широким спектром клінічних наслідків, зумовлених тиреотоксикозом периферичних тканин унаслідок підвищеної концентрації трийодтироніну (Т3) у сироватці крові (табл. 1).

Осіб з дефіцитом МКТ-8 виявляють за допомогою прицільного секвенування гена SLC16A2, розміщеного на X-хромосомі. Секвенування проводять вибраним пацієнтам (переважно чоловічої статі) з відповідними клінічними та біохімічними характеристиками; також застосовується секвенування екзому. Встановлення точного діагнозу дефіциту МКТ-8 потребує виявлення відомої патогенної мутації.
Лікування дефіциту МКТ-8 має бути спрямоване на збільшення впливу тиреоїдних гормонів на мозок, який перебуває в стані гіпотиреозу, та на усунення тиреотоксикозу периферичних тканин. Із цією метою застосовуються левотироксин (як монотерапія або в поєднанні з пропілтіоурацилом – ПТУ) й аналоги T3 – дийодтиропропіонова кислота (ДЙТПК) і трийодтирооцтова кислота (ТЙТОК), однак жоден з наявних методів лікування не здатен повністю вилікувати пацієнтів з нейрокогнітивним фенотипом; такі хворі потребують підтримувальної/симптоматичної терапії.
Діагностика
- За підозри на дефіцит МКТ-8 рекомендовано провести повну клінічну оцінку для виявлення відомих клінічних фенотипів хвороби, а також фізикальне обстеження, включаючи неврологічні методи.
- Також рекомендовано додаткові обстеження, зокрема визначення вільного Т3 (вТ3), вільного тироксину (вТ4), зворотного T3 (зТ3) і тиреотропного гормону (ТТГ) з інтерпретацією результатів у контексті специфічних для конкретного віку референсних інтервалів.
- Секвенування SLC16A2 рекомендовано проводити в усіх пацієнтів чоловічої статі з такими великими критеріями, як типовий біохімічний профіль (нормальний або дещо підвищений ТТГ, низький або низький нормальний вТ4, підвищений вТ3, низький зT3, підвищене співвідношення вT3/вT4 або T3/зT3) у поєднанні з глобальною затримкою розвитку та гіпомієлінацією на МРТ, клінічними ознаками порушень руху (дистонія, брадикінезія), стійкими примітивними рефлексами або сімейним анамнезом дефіциту MКT-8.
- Розглянути доцільність секвенування SLC16A2 слід також у всіх пацієнтів з типовими результатами лабораторних досліджень (нормальний або дещо підвищений ТТГ, низький або низький нормальний Т4, підвищений Т3, низький зT3, підвищене співвідношення вT3/вT4 або T3/зT3) та незначною затримкою розвитку чи поведінковими змінами.
- Пренатальне секвенування SLC16A2 (за допомогою біопсії ворсин хоріону або амніоцентезу) слід розглянути в разі вагітності плодом чоловічої статі за наявності сімейного анамнезу дефіциту MКT-8 і ризику наявності мутантного алеля в плода відповідно до генетичної сегрегації.
Лікування
- Постнатальна монотерапія левотироксином не рекомендована.
- Рекомендовано лікування ТЙТОК або ДЙТПК.
- Дозу ТЙТОК або ДЙТПК слід титрувати до цільових показників сироваткового T3 у межах 1,4-2,5 нмоль/л з метою зменшення периферичного тиреотоксичного стану, якщо не виникають дозозалежні побічні ефекти. Вища доза ТЙТОК може зменшити вміст T3 до нижньої межі або нижче меж норми для конкретного віку, маючи потенційно сприятливий вплив на нейророзвиток.
- Усім пацієнтам рекомендовано довготривалу терапію ТЙТОК або ДЙТПК (якщо не виникають побічні ефекти або непереносимість).
- Якщо аналоги тиреоїдних гормонів недоступні, слід розглянути застосування комбінації левотироксину та ПТУ.
- Враховуючи рідкісні, але потенційно небезпечні та загрозливі для життя побічні ефекти ПТУ й невизначені довгострокові впливи аналогів тиреоїдного гормону, слід ретельно зважити ризики та переваги застосування цих препаратів, особливо в пацієнтів з порушеною функцією печінки.
- Усім пацієнтам з дефіцитом МКТ-8 рекомендовано перебувати під наглядом (дитячого) ендокринолога та (дитячого) невролога.
- Випадки дефіциту МКТ-8 рекомендовано вести у складі багатодисциплінарної команди, яка включає (дитячого) ендокринолога, невролога, кардіолога, дієтолога, ерготерапевта, логопеда, фізіотерапевта, реабілітолога, ортопедичного хірурга та працівників спеціалізованого дитячого садка. Загальний нагляд за роботою команди має здійснювати індивідуальний менеджер кожного клінічного випадку.
- Рекомендовано ретельно оцінювати харчування пацієнта з метою підтримки маси тіла, адекватної для конкретного віку, та запобігати недостатній вгодованості. Для забезпечення надходження належної кількості калорій і профілактики мальнутриції пропонується запровадити харчування через перкутанний ентеральний зонд і провести консультацію дієтолога.
- Усім пацієнтам слід запропонувати емпіричне симптоматичне лікування неврологічних симптомів, включаючи дистонію/гіпертонію (спазмолітичні препарати), слинотечу (антихолінергічні препарати), істинну епілепсію (її треба диференціювати з пароксизмальними руховими розладами), а також скеровувати пацієнтів до реабілітологів і хірургів-ортопедів для профілактики контрактур, сколіозу та підвивиху стегна.
- Часті шлунково-кишкові проблеми (гастроезофагальний рефлюкс та/або гастропарез, закреп) рекомендовано лікувати відповідно до стандартних рекомендацій.
Дефіцит селенопротеїну
Вступ
Селен є важливим слідовим елементом, що у складі амінокислоти селеноцистеїну виступає компонентом 25 різних селенопротеїнів, включаючи фермент йодтироніндейодиназу. Низка гомо- та гетерозиготних мутацій гена SECISBP2 здатна перешкоджати інкорпорації селеноцистеїну в ці білки в ході синтезу останніх, що асоціюється з розладами функції щитоподібної залози (ЩЗ) та низьким рівнем селену плазми крові. Дефіцит селенопротеїнів і спричинене нестачею антиоксидантних селеноферментів окисне ушкодження зумовлюють різні фенотипи цієї патології. Дефекти іншого фактора цього біосинтетичного шляху – синтази SEPSECS – призводять до прогресивної мікроцефалії з нормальними рівнями T4 та селену в сироватці крові.
Діагностика
Дефіцит селенопротеїнів спричиняє мультисистемне захворювання (табл. 2).
- У більшості випадків відзначаються низький зріст і затримка розвитку. Також найпоширеніший фенотип передбачає слабкість і гіпотонію у зв’язку з прогресивною дегенерацією певних груп м’язів (кравецького, адукторів, осьових параспінальних). Інші фенотипи характеризуються аневризматичною дилатацією грудного відділу аорти, сенсоневральною глухотою, фоточутливістю шкіри та неплідністю (в чоловіків).
- Терапія ліотироніном корегує субнормальний уміст вT3 та покращує лінійний ріст у разі призначення у формі монотерапії або в комбінації з соматотропіном, однак особи, які не лікуються, також можуть досягти нормального зросту. Хоча пероральні добавки селену при дефіциті SECISBP2 відновлюють концентрацію селену в плазмі крові, вони не здатні скорегувати дефіцит циркулювального селенопротеїну або порушення конверсії T4 у T3. Лікування антиоксидантами, наприклад α-токоферолом, захищає клітини від окисного пошкодження, не впливаючи негативно на метаболічний фенотип.

Діагностика
- Рекомендовано проводити біохімічний аналіз крові. Типовими ознаками дефіциту селенопротеїну внаслідок мутацій SECISBP2 або TRU-TCA1-1 є підвищений вT4, нормальний або низький вT3, підвищений зT3 та низька концентрація селену.
- При встановленні генетичного діагнозу рекомендовано секвенування всього екзому або геному з виявленням інтронних мутацій SECISBP2.
Лікування
- Рекомендовано відстежувати затримку росту та розвитку в дитинстві.
- Рекомендовано періодично проводити МРТ й у вибраних випадках біопсію м’язів, визначення життєвої ємності легень і полісомнографію.
- Рекомендовано спостерігати за пацієнтами щодо прогресивної сенсоневральної глухоти й аневризм грудного відділу аорти, а також фоточутливості шкіри та неплідності (в чоловіків).
- Пацієнтам із субнормальною концентрацією вТ3 рекомендовано призначати ліотиронін.
- Добавки селену при дефіциті SECISBP2 не рекомендовані.
- Можна розглянути лікування антиоксидантами (α-токоферолом).
Дефекти йодтироніндейодинази
Вступ
Мутації генів йодтироніндейодинази є дуже рідкісними; у світі зареєстровано лише три родини з цими мутаціями. У членів цих родин спостерігаються підвищений рівень зТ3 та підвищення відношення зТ3 до загального Т3. Специфічне лікування таких станів наразі не визначене. Імовірно, поєднання цих мутацій з іншими вродженими вадами ЩЗ потребує призначення замісної терапії левотироксином.
Діагностика
- Передусім рекомендовано задокументувати підвищений уміст вT3 та збільшене співвідношення зT3/T3 за відсутності нетиреоїдної патології.
- Оскільки тип успадкування є домінантним, доцільно обстежити членів сім’ї, насамперед батьків пацієнта.
- Ключове значення в діагностиці має виявлення мутації гена DIO1.
Чіткі рекомендації щодо лікування наразі відсутні.
Диференційна діагностика підвищеного вмісту гормонів ЩЗ у поєднанні з відсутністю супресії ТТГ
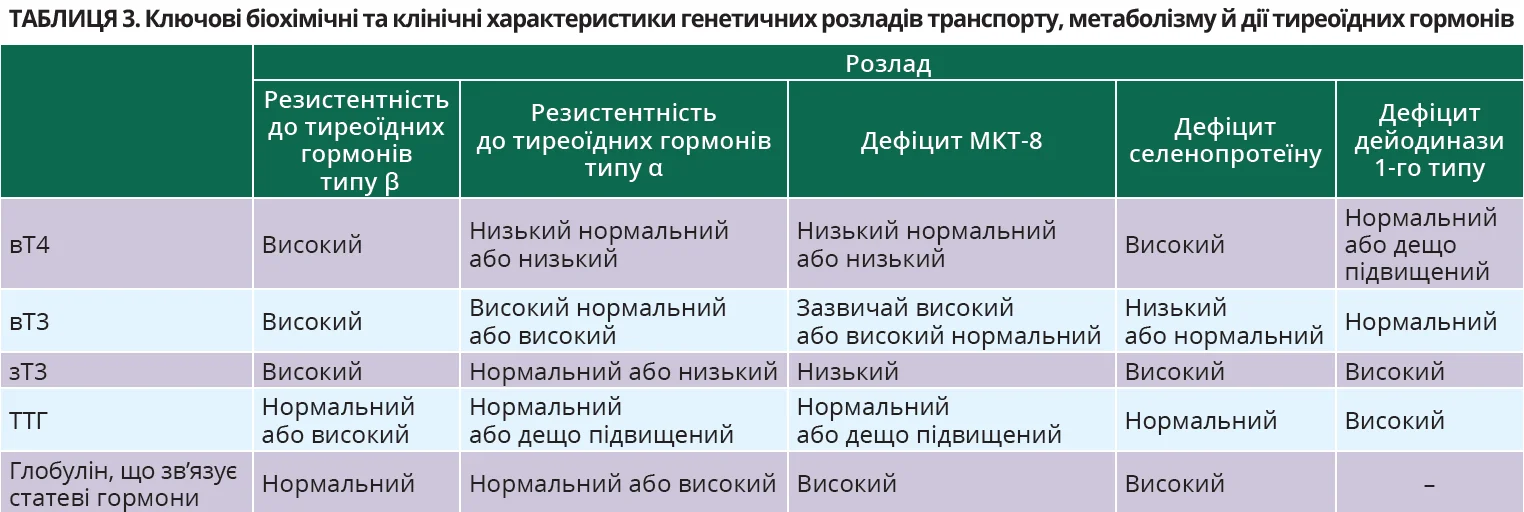
Перевищення референсного показника тиреоїдних гормонів і відсутність супресії ТТГ є одним з найскладніших патернів обстежень для диференційної діагностики. У такій ситуації першим кроком має бути виключення потенційних нетиреоїдних причин і вживання певних медикаментів (аміодарону, тироксину). Після цього необхідно виключити артефакти та помилки діагностичної лабораторії. Якщо ж було підтверджено істинну гіпертироксинемію на тлі відсутності пригнічення ТТГ, призначаються додаткові обстеження: вT3 з розрахунком співвідношення T4/T3, зT3, тиреоїдні автоантитіла, глобулін, що зв’язує статеві гормони (табл. 3).
Література
Persani L., Rodien P., Moran C., Edward Visser W., Groeneweg S., Peeters R., Refetoff S., Gurnell M., Beck-Peccoz P., Chatterjee K. 2024 European Thyroid Association Guidelines on diagnosis and management of genetic disorders of thyroid hormone transport, metabolism and action. Eur. Thyroid J. 2024; 13 (4): e240125. doi: 10.1530/ETJ-24-0125.