Огляд ендокринної гіпертензії
Підготувала Ольга Королюк
За підрахунками, кожен третій житель планети може мати артеріальну гіпертензію (АГ). Підвищення артеріального тиску (АТ) – одна з найчастіших причин звернення до лікаря й основний чинник ризику ішемічної хвороби серця, серцевої недостатності й інсульту. Поширеність АГ зростає з віком, коли здебільшого діагностують первинну (есенціальну) АГ. Вторинна АГ найчастіше зумовлена хворобами нирок та ендокринними розладами.
Левову частку ендокринних АГ становлять хвороби надниркових залоз, як-от первинний альдостеронізм (ПА), надлишок глюкокортикоїдів, синдроми феохромоцитоми (ФХЦ) та парагангліоми. Синдром Хрусоса – рідкісна хвороба, що характеризується вродженою гіперплазією надниркових залоз, АГ та резистентністю до глюкокортикоїдів. Серед інших ендокринних причин АГ варто вказати надлишок або дефіцит гормону росту, дисфункцію щитоподібної залози, первинний гіперпаратиреоз, дефіцит тестостерону, ожиріння, резистентність до інсуліну, метаболічний синдром і дефіцит вітаміну D. Діагностика ендокринної АГ здебільшого полягає у виявленні гіперпродукції певного гормону та визначає тактику лікування – специфічна фармакотерапія або хірургічне втручання, що може сприяти повному одужанню й уникненню потреби довічної антигіпертензивної терапії.
Як виміряти АТ?
Золотим стандартом залишається ручне вимірювання ртутним сфігмоманометром за допомогою стетоскопа. Через екологічні проблеми, пов’язані з ртуттю, від цієї техніки відмовляються, віддаючи перевагу автоматичним пристроям. Проте валідність результатів автоматичних вимірювань у клінічній практиці обмежена через значні похибки. Пристрій має бути перевіреним і відкаліброваним, а манжета має бути правильного розміру (75-100% плеча). АТ оцінюється за середнім значенням принаймні двох правильно виміряних показників у положенні сидячи, здійснених під час принаймні двох офісних візитів. Найкраще, коли 3-5 хвилин перед вимірюванням пацієнт проведе в спокої, лежачи або зручно сидячи без перехрещування ніг, не розмовляючи та не рухаючись. Бажано уникати вживання кофеїну, куріння та фізичних вправ перед вимірюванням. Одяг, який закриває манжету на плечі, варто зняти. Під час першого візиту записується АТ на обох руках, ураховуючи вищі показники з двох вимірювань, проведених з інтервалом 1-2 хвилини. Під час вимірювання рука пацієнта повинна мати опору з положенням надпліччя на рівні правого передсердя. Перший тон Короткова відповідає показнику систолічного АТ (САТ), останній чутний звук – показнику діастолічного АТ (ДАТ).
Класифікація гіпертензії
На сьогодні у світі існують різні визначення АГ та класифікації АТ у дорослих пацієнтів віком від 18 років (табл. 1).

Поширеність ендокринної гіпертензії
За сучасними даними, понад 5-10% усіх випадків гіпертензії зумовлено ендокринною патологією, найчастіше гіперпродукцією мінералокортикоїдів (первинний гіперальдостеронізм), катехоламінів (ФХЦ), глюкокортикоїдів (синдром Кушинга) та гормонів щитоподібної залози. Найпоширеніші причини вторинних АГ підсумовано в таблиці 2.

Поширеність і типові причини резистентної АГ
Резистентну АГ визначають як недосягнення цільових рівнів АТ, незважаючи на застосування ≥3 антигіпертензивних препаратів (АГП). Після 5-річного спостереження за учасниками дослідження ALLHAT установлено, що 34% не досягли контролю АТ при застосуванні ≥2 АГП. Резистентна АГ у молодих пацієнтів завжди має спонукати до ретельного пошуку вторинних причин: ФХЦ, синдром Кушинга, гіпер- або гіпотиреоз, акромегалія, інсулінорезистентність.
У всіх випадках варто розпитати пацієнта про харчові звички (споживання солі), коливання маси тіла, використання рекреаційних засобів, оральних контрацептивів, безрецептурних ліків і харчових добавок, включаючи чаї та трав’яні збори; зібрати докладний сімейний анамнез.
Огляд систем і об’єктивне обстеження треба проводити з акцентом на ознаки та симптоми вторинної АГ й ознаки ураження органів-мішеней. Наприклад, симптоми ФХЦ та гіпертиреозу включають головний біль, серцебиття, напади тривоги й рясне потовиділення. Раніше тріада «головний біль, серцебиття та пітливість» у пацієнта з АГ вважалася типовою характеристикою ФХЦ (чутливість – 91%, специфічність – 94%).Проте, за даними останніх досліджень, ця тріада трапляється лише в 10% випадків, натомість ≥10% пацієнтів можуть мати нормальний тиск без будь-яких симптомів.
Первинний гіперальдостеронізм проявляється гіпертензією від легкої до тяжкої; можлива гіпокаліємія, що проявляється поліурією, міопатією й аритмією. Утім, існують нормокаліємічний і нормотензивний варіанти.
Пацієнти із синдромом Кушинга часто скаржаться на збільшення маси тіла, безсоння, депресію, втому, легке утворення синців, акне та гірсутизм у жінок; але на ранніх етапах ці симптоми проблематично відрізнити від інших форм ожиріння або погано контрольованого діабету.
Проявами інсулінорезистентності є надмірна маса тіла, порушення регуляції глюкози, acanthosis nigricans.
Низький рівень реніну асоціюється з декількома причинами АГ (табл. 3).

Незважаючи на значний прогрес у розумінні патофізіології АГ, контроль тиску залишається складним і далеким від оптимального. Нещодавні масштабні метааналізи та дослідження генотипу виявили деякі «гени ризику» АГ. Зокрема, низькочастотний нонсенс-варіант у гені ENPEP (кодує фермент амінопептидазу А, що перетворює ангіотензин II на ангіотензин III), а також рідкісні й низькочастотні місенс-варіанти в генах NPR1, DBH та PTPMT1. Ген DBH кодує фермент дофамін-β-гідроксилазу (каталізує перетворення дофаміну на норадреналін) і впливає на вегетативну нервову систему. Ген PTPMT1 кодує мітохондріальний білок тирозинфосфатазу-1, який впливає на продукцію інсуліну.
Клінічна діагностика ендокринної гіпертензії
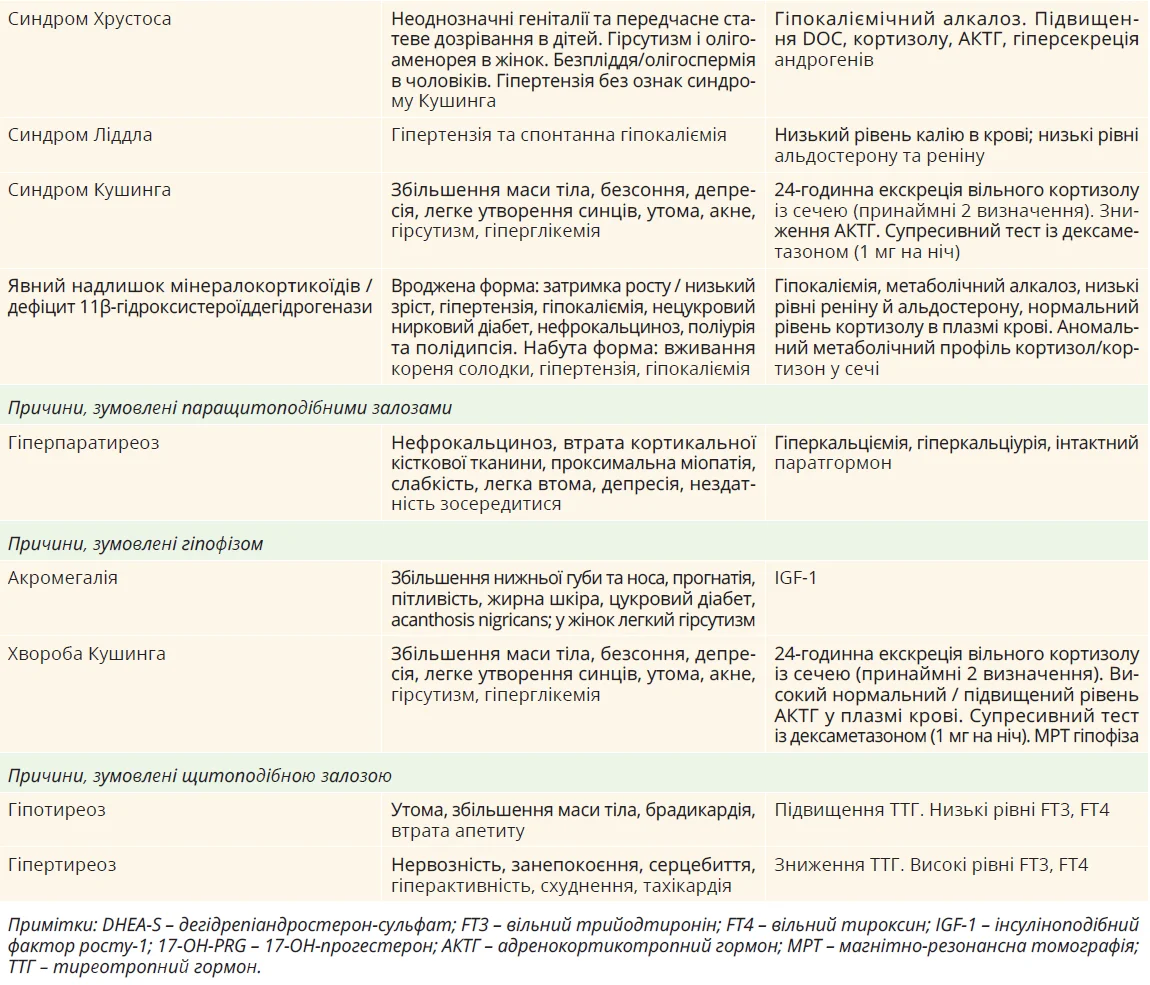
Обстежуючи пацієнта з підозрою на ендокринну АГ, треба виключити інші вторинні причини, насамперед хвороби нирок. Слід з’ясувати початок АГ, відповідь на попередню антигіпертензивну терапію та дотримання пацієнтом режиму прийому АГП; ретельно вивчити історію ураження органів-мішеней (ретинопатію, нефропатію, переміжну кульгавість, захворювання серця, сонних артерій і черевної порожнини) й оцінити загальний статус серцево-судинного ризику. Клінічний анамнез, результати фізикального обстеження та рутинних лабораторних досліджень, які дають змогу припустити специфічні ендокринні причини АГ, підсумовано в таблиці 4.

Первинний альдостеронізм
Поширеність
За попередніми даними, поширеність ПА становить 1-2% усіх випадків АГ, проте нові дані вказують значно більші цифри (>5%, можливо >10%). У пацієнтів із легкою-помірною АГ без гіпокаліємії поширеність ПА становить 3%; у пацієнтів із резистентною АГ – 17-23%. У дослідженні за участю 1616 пацієнтів із резистентною АГ співвідношення альдостерон/ренін >65 із концентрацією альдостерону в плазмі >416 пмоль/л (15 нг) виявлено у 21% (n=338); після супресивного сольового тесту діагноз ПА підтверджено лише в 11% (n=182).
У пацієнтів з інциденталомою надниркових залоз і АГ поширеність ПА становить 2%. Близько 63% пацієнтів із ПА можуть мати нормальні рівні калію крові. Гіпертензію з низьким рівнем реніну не завжди легко відрізнити від ПА. У дослідженні за участю 553 пацієнтів із ПА гіпокаліємію мали 56%, супутні серцево-судинні та цереброваскулярні хвороби – 16%.
Отже, скринінг на ПА рекомендовано в таких групах пацієнтів:
- резистентна АГ;
- гіпокаліємія (спричинена діуретиками, спонтанна);
- сімейним анамнез ранньої АГ або порушення мозкового кровообігу в молодому віці;
- АГ й інциденталома надниркових залоз, виявлена при візуалізації.
Етіологія
Існують спорадичні та сімейні форми ПА. Багато спорадичних випадків ПА спричинені аденомами наднирників, що продукують альдостерон. Іншою доволі частою причиною є двобічна гіперплазія клубочкової зони кори наднирників. Ці стани важливо диференціювати через різні підходи до лікування: адреналектомія при аденомі, антагоністи альдостерону при гіперплазії. У рідкісних випадках причинами ПА є карцинома чи однобічна гіперплазія кори наднирників, відома як первинна гіперплазія надниркових залоз.
Загальна поширеність сімейних форм ПА становить ≈2% усіх випадків. Генетичні та клінічні характеристики сімейних форм ПА підсумовано в таблиці 5.

Сімейний ПА 1 типу зумовлений автосомно-домінантним успадкуванням дефекту гібридного гена CYP11B1/CYPB2 у хромосомі 8, який кодує 11-β-гідроксилазу/альдостеронсинтазу. Як наслідок, виникає ектопічна експресія активності альдостеронсинтази в сітчастій зоні (zona fasciculata), яка продукує кортизол; продукція мінералокортикоїдів регулюється кортикотропіном. Рівні гібридних стероїдів 18-оксокортизолу (18-oxoF), 18-гідроксикортизолу (18-OHF) у сечі в ≈20 разів перевищують рівні, які спостерігаються за спорадичних форм ПА. Клінічними ознаками хвороби є внутрішньочерепні аневризми та геморагічний інсульт. Діагноз установлюють шляхом підтвердження пригнічення сироваткового альдостерону дексаметазоном за допомогою тесту Ліддла (зниження рівня альдостерону в плазмі <4 нг/дл під впливом дексаметазону в режимі 0,5 мг що 6 годин протягом 48 годин) або шляхом генетичного тестування (Саузерн-блот або полімеразна ланцюгова реакція).
Сімейний ПА 2 типу спричинений мутаціями хлоридного каналу з внутрішнім випрямленням CLCN2. На відміну від 1 типу не чутливий до лікування глюкокортикоїдами.
Сімейний ПА 3 типу зумовлений гетерозиготною мутацією посилення функції в калієвому каналі GIRK4 (кодується KCNJ5), що призводить до посилення експресії альдостеронсинтази та гіперпродукції альдостерону. Пацієнтам із сімейним ПА 3 типу притаманні атрофія клубочкової зони та дифузна гіперплазія пучкової зони кори наднирників, тяжка АГ із дитинства (вік ≈7 років), часто резистентна до медикаментозної терапії, парадоксальне підвищення рівня альдостерону після введення дексаметазону та продукція 18-oxoF і 18-OHF у 10-1000 разів вища, ніж у пацієнтів із сімейним ПА 1 та 2 типів або спорадичним ПА. Здебільшого такі випадки потребують двобічної адреналектомії.
Сімейний ПА 4 типу виникає внаслідок мутацій зародкової лінії в субодиниці кальцієвого каналу Т-типу в гені CACNA1H.
У пацієнтів із ПА виявляються мутації зародкової лінії в CACNA1D, який кодує субодиницю L-типу потенціалозалежного кальцієвого каналу CaV1.3. Іноді такі мутації пов’язані із судомами та неврологічними відхиленнями.
Діагностика – скринінг і підтверджувальні тести
Діагноз ПА потребує вимірювання рівня альдостерону в плазмі крові (PA) й активності реніну (PRA) чи концентрації реніну. Виміряти PRA доволі складно, кількісне визначення перетворення ангіотензиногена на ангіотензин під впливом реніну виконується за допомогою радіоімунних аналізів, не стандартизованих між лабораторіями. Можливе також безпосереднє вимірювання молекул реніну в плазмі крові методом автоматизованого хемілюмінесцентного імунологічного аналізу. Діагностичним критерієм ПА є співвідношення PA/PRA >30 за умови, що PA >20 нг/дл (чутливість – 90%, специфічність – 91%).
Перед подальшим діагностичним обстеженням слід відкорегувати гіпокаліємію, котра знижує секрецію альдостерону. Діагностувати ПА в пацієнтів, які лікуються інгібітором ангіотензинперетворювального ферменту (іАПФ), блокатором рецепторів ангіотензину ІІ (БРА), блокатором кальцієвих каналів (БКК) або діуретиком (усі засоби підвищують PRA та знижують PA/PRA), можна лише тоді, коли рівень реніну знижений, а рівень PA підвищений до двоцифрового значення.
Хибнопозитивне підвищення PA/PRA можливе в жінок під час лютеїнової фази менструального циклу, а також у тих, хто приймає контрацептиви зі вмістом естрогенів. Для виключення медикаментозних впливів рекомендується скасувати β-блокатори, іАПФ, БРА, інгібітори реніну, дигідропіридинові БКК, нестероїдні протизапальні препарати й α2-агоністи центральної дії приблизно за 2 тижні до визначення PA/PRA, а сечогінні засоби (спіронолактон, еплеренон, амілорид, тріамтерен, петльові діуретики) та корінь солодки – за 4 тижні до визначення PA/PRA.
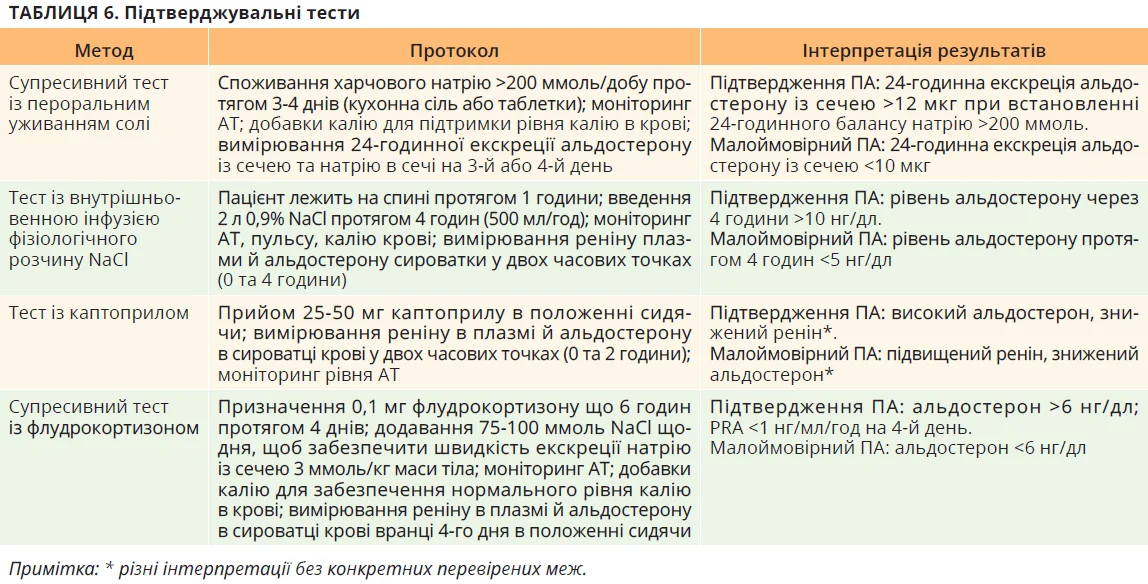
Існує багато методів підтверджувальних тестів, які підсумовано в таблиці 6.
Нещодавнє дослідження за участю 148 пацієнтів з АГ виявило, що нічний діагностичний тест із фармацевтичною блокадою ренін-ангіотензин-альдостеронової системи (РААС) дексаметазоном, каптоприлом і валсартаном має чутливість 98%, специфічність 100% для діагностики ПА; тест швидкий, безпечний, простий у виконанні та має низьку вартість (Tsiavos V. et al., 2016).
Локалізація
Для диференціації гіперплазії й аденоми використовують комп’ютерну томографію (КТ) і МРТ. Якщо планується хірургічне втручання, можна виконати забір зразків венозної крові з надниркової вени (AVS) з інфузією косинтропіну (АКТГ). Для виявлення однобічної аденоми методом AVS визначається «кортизол-кореговане» співвідношення альдостерону (значення альдостерон/кортизол на боці аденоми, поділене на значення нормального відношення альдостерон/кортизол надниркових залоз); показник >4 вказує на однобічну аденому. Виявлення однобічних аномалій при AVS і КТ/МРТ надниркових залоз становить 60,5 та 56% відповідно.
Медикаментозне лікування
Рекомендації клінічної практичної настанови Ендокринного товариства (2016) щодо лікування ПА: пацієнтам з АГ, спонтанною гіпокаліємією, концентрацією альдостерону в плазмі >20 нг/дл (550 пмоль/л) і рівнем реніну нижче межі визначення можна не проводити подальших підтверджувальних тестів; виконати візуалізацію та/або AVS; за неможливості або небажання проведення адреналектомії лікувати за допомогою АМР.
Для лікування двобічної гіперплазії застосовують спіронолактон, еплеренон та/або амілорид. Спіронолактон – неселективний конкурентний АМР, вважається терапією першої лінії в дозах від 12,5 до 400 мг/добу (зазвичай 12,5-50 мг/добу); діє також як антагоніст рецепторів андрогенів, слабкий антагоніст рецепторів глюкокортикоїдів і агоніст рецепторів прогестерону, що зумовлює дозозалежні побічні ефекти: гіперкаліємію, гіпонатріємію, гінекомастію, порушення менструального циклу, чутливість грудей і зниження лібідо в жінок, гінекомастію в чоловіків.
Еплеренон – селективний АМР із меншою антиандрогенною дією, але й меншою спорідненістю до мінералокортикоїдних рецепторів і менш потужним антигіпертензивним ефектом порівняно зі спіронолактоном. Досягнення аналогічного до спіронолактону ефекту зазвичай потребує призначення вищих доз еплеренону – 25-50 мг двічі на день.
У разі сімейного гіперальдостеронізму 1 типу використовується дексаметазон, що пригнічує гіперпродукцію альдостерону через супресію АКТГ.
Хірургічне лікування
Видалення альдостерон-продукувальної аденоми надниркових залоз покращує контроль АТ практично в усіх пацієнтів (рис. 1); близько 60% пацієнтів виліковуються, тобто досягають нормалізації АТ без АГП. На цей результат впливають різні чинники, включаючи вік, тривалість АГ, супутню ниркову недостатність, використання ≥2 АГП до операції, сімейний анамнез АГ тощо. Що стосується післяопераційних показників чутливості до інсуліну, то дані літератури суперечливі – від цілковитої нормалізації до відсутності істотної різниці до та після операції.
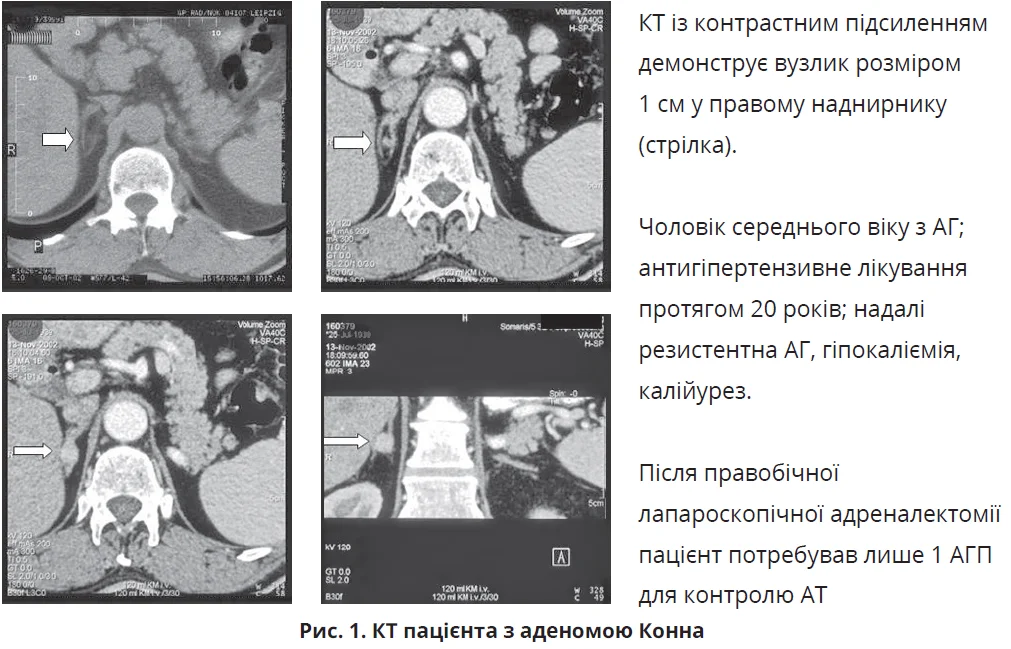
За однобічної альдостерономи/гіперплазії показане хірургічне втручання – адреналектомія.
У віці понад 40 років збільшується ризик виникнення інциденталоми наднирників. Однобічна адреналектомія іноді корисна за двобічної гіперплазії надниркових залоз. Якщо пацієнт відмовляється від операції, проводять медикаментозне лікування.
Феохромоцитома / симпатична парагангліома
Поширеність
Це рідкісні пухлини, що трапляються в 0,2% пацієнтів з АГ. Щорічна захворюваність на ФХЦ становить майже 0,8 на 100 тис. людино-років. Може виникати в будь-якому віці, але зазвичай упродовж четвертої-п’ятої декади життя.
Етіологія
Це нейроендокринні пухлини, які складаються з хромафінної тканини, що містить нейросекреторні гранули. На сьогодні ≈40% пацієнтів із ФХЦ / симпатичною парагангліомою (СПГ) мають мутацію зародкової лінії, незалежно від віку появи пухлини та сімейного анамнезу. Наразі відомо 10 клінічно значущих синдромів, асоційованих із ФХЦ/СПГ: множинна ендокринна неоплазія (MEN) 2 типу, синдром фон Гіппеля – Ліндау (VHL), нейрофіброматоз 1 типу, синдроми парагангліоми 1-5, спричинені мутаціями генів сукцинатдегідрогенази, SDHD (синдром 1), SDHAF2 (синдром 2), SDHC (синдром 3), SDHB (синдром 4) та SDHA (синдром 5), а також синдроми спадкової ФХЦ внаслідок мутацій зародкової лінії в генах, що кодують трансмембранний білок-127 (TMEM127) і фактор X, асоційований із MYC (MAX). Інші гени сприйнятливості включають EGLN1 (PHD2), EGLN2 (PHD1), DNMT3A, IDH1, FH, MDH2, SLC25A11, KIF1B та HIF2A. Чітких вказівок щодо терміну проведення генетичного тестування в цій когорті не встановлено з огляду на економічну ефективність.
Клінічні особливості
Клінічна картина пацієнтів із ФХЦ/СПГ дуже варіабельна: від відсутності / незначних симптомів до серйозних проявів, загрозливих для життя.
За відсутності симптомів об’ємні утворення виявляють випадково внаслідок обстеження з інших причин. Близько 15% пацієнтів із ФХЦ мають нормальний АТ. Класична тріада «стукальний головний біль, сильне потовиділення й серцебиття» виникає спорадично; симптоми тривають від кількох хвилин до 1 години. Пароксизмальна АГ трапляється в 35-50% хворих; між епізодами пацієнти зазвичай не мають симптомів. У пацієнтів із ФХЦ немає кореляції між рівнем АТ і рівнями катехоламінів у крові. Проте високі стрибки АТ й інші симптоми пов’язані з неконтрольованим вивільненням катехоламінів, зумовлюють високу поширеність серцево-судинних невідкладних станів (інфаркт міокарда, інсульт, гостра серцева недостатність) і гострий початок кардіоміопатії такоцубо. Пацієнти із синдромом MEN2 і VHL можуть мати клінічно «німі» ФХЦ.
Діагностика – скринінг
Діагноз установлюють шляхом вимірювання метанефринів (метанефрин і норметанефрин) у плазмі або сечі. Якщо вільні метанефрини в плазмі неможливо виміряти методом високоефективної рідинної хроматографії (HPLC) або високопродуктивної автоматизованої рідинно-хроматографічної тандемної мас-спектрометрії (LC-MS/MS), можливе визначення вільних метанефринів у плазмі крові методом радіоімунологічного аналізу чи хромограніну А в плазмі крові. У рідкісних випадках ФХЦ вивільняє великий O-метильований метаболіт дофаміну метокситирамін, рівень якого підвищений за позанаднирникової локалізації пухлини (парагангліоми шиї й основи черепа) та в разі метастатичного захворювання.
У пацієнтів із нирковою недостатністю концентрація вільних метанефринів у плазмі крові підвищується в декілька разів. Для оптимальної діагностики краще використовувати встановлені референтні значення вільних метанефринів у плазмі крові та 24-годинних фракціонованих метанефринів у сечі відповідно до віку та статі. Верхній граничний рівень вільного норметанефрину в плазмі вищий у літніх пацієнтів.
Причиною хибнопозитивних результатів біохімічних тестів може бути застосування певних ліків (табл. 7).

Визначення вільних метанефринів у плазмі крові й О-метильованого метаболіту дофаміну метокситираміну не потребує жодних харчових обмежень, за винятком нічного голодування.
Настанова з клінічної практики Ендокринного товариства (2014) рекомендує генетичне тестування всім пацієнтам із ФХЦ/СПГ за умови спільного прийняття рішення з пацієнтом. Пацієнтам із СПГ показане тестування на мутації сукцинатдегідрогенази (SDH), пацієнтам із метастатичним захворюванням – на мутації SDHB. Слід зазначити, що SDHD та SDHAF2 мають материнський імпринтинг, що пропускає одне або декілька поколінь. У віці до 20 років найпоширеніші спадкові синдроми ФХЦ/СПГ пов’язані з VHL, синдромом парагангліоми 4 типу та нейрофіброматозом 1 типу. У віці від 30 до 50 років ФХЦ найчастіше пов’язана з MEN 2 типу. Пенетрантність ФХЦ/СПГ в осіб із мутацією зародкової лінії RET становить у середньому 50% до 44 років.
Метастатичні форми ФХЦ/СПГ становлять близько 10% пухлин наднирників і приблизно 35% екстра-надниркових пухлин. Ризик метастазування вищий для пухлин розміром понад 5 см і за наявності мутації зародкової лінії гена SDHB.
Локалізація
Локалізацію близько 95% пухлин можна встановити за допомогою КТ або МРТ. У разі метастатичних ФХЦ краще проводити позитронно-емісійну томографію з 18F-фтордопаміном і 18F-FDG, аніж сцинтиграфію з 123I-MIBG або 131I-MIBG. Перед проведенням 123I-MIBG/131I-MIBG-сканування потрібно припинити вживання ліків, які перешкоджають захопленню контрасту (БКК, антипсихотичні засоби).
Настанови Ендокринного товариства 2014 р. рекомендують використання 123I-MIBG у пацієнтів із метастатичною ФХЦ/СПГ, коли планується променева терапія 131I-MIBG, або за високого ризику метастатичної хвороби (великий розмір пухлини, позанадниркова пухлина, мультифокальна локалізація чи рецидив). Серед пацієнтів із СПГ голови та шиї добру чутливість має 111In-октреотид. Чудову роздільну здатність виявлення ФХЦ/СПГ мають сучасні функціональні методи візуалізації, як-от мічена 68Ga 1,4,7,10-тетраазацилододекан-1,4,7,10-тетраоцтова кислота-октреотат (DOTATATE) міченого 18F L-дигідроксифенілаланіну (L-DOPA).
Медикаментозне лікування
Ендокринне товариство, Американська асоціація клінічної хімії та Європейське товариство ендокринології опублікували настанови з клінічної практики, які рекомендують передопераційну блокаду гормонально-функціональних ФХЦ/СПГ для запобігання серцево-судинним ускладненням, АГП та засоби для нормалізації частоти серцевих скорочень.
Для передопераційної підготовки використовують селективні α-адреноблокатори (доксазозин [Cardura], празозин [Minipress] або теразозин [Hytrin]) із подальшим призначенням β-адреноблокаторів (пропранолол, атенолол). Рекомендовано дієту з високим умістом натрію та достатнє споживання рідини, щоб запобігти зниженню АТ після операції. Близько 50% пацієнтів із метастатичною ФХЦ відповідають на терапію 131I-MIBG частковою ремісією або стабілізацією хвороби. При метастатичних формах також тривало використовують селективні α-блокатори. Нові варіанти терапії метастатичної ФХЦ/СПГ включають 90Y-DOTATATE та 177Lu-DOTATATE. Хіміотерапія зазвичай проводиться за протоколом Авербуха. Нові методи лікування пропонують інгібітори тирозинкінази в окремих пацієнтів.
Хірургічне лікування
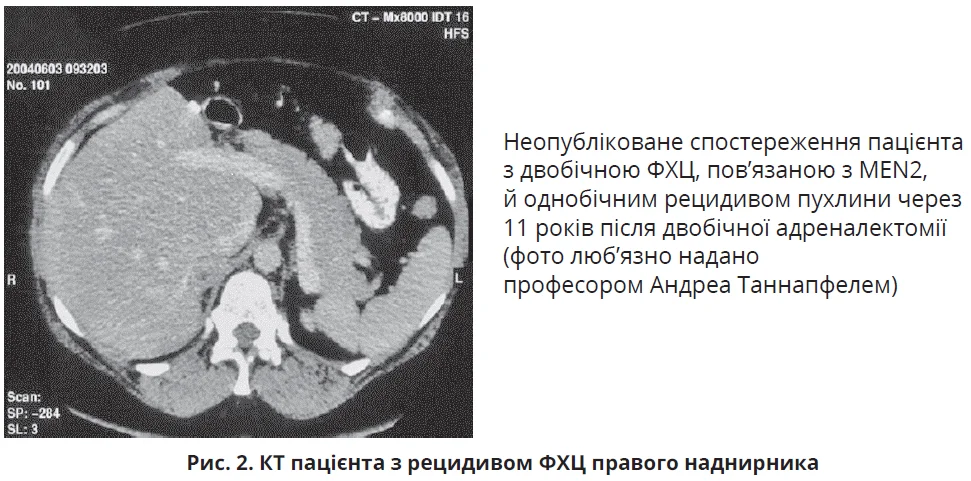
Для забезпечення повної резекції пухлини, запобігання розриву капсули й уникнення місцевого рецидиву відкрита адреналектомія вважалася кращою процедурою видалення пухлини розміром понад 5 см порівняно з лапароскопічною або ретроперитонеальноскопічною резекцією (рис. 2 та 3). Проте нещодавнє багатоцентрове когортне дослідження за участю 625 пацієнтів із двобічною ФХЦ розміром до 6 см, які лікувалися в період 1950-2018 рр., порівнювало наслідки лікування методом тотальної адреналектомії та кортикально-зберігальної адреналектомії. Після кортикально-зберігальної адреналектомії не спостерігалося зниження виживаності, незважаючи на рецидив пухлини в 13% випадків. Автори рекомендують розглянути можливість кортикально-зберігальної адреналектомії в усіх пацієнтів зі спадковою ФХЦ.

Багатоцентрове міжнародне ретроспективне дослідження за участю пацієнтів різного віку з варіантом Met918Thr RET, які спостерігалися в період 1970-2016 рр., виявило, що зберігальна операція при MEN типу 2B може зберегти нормальну функцію надниркових залоз. Операцію зі збереженням надниркових залоз проведено 31 пацієнту; в 3 пацієнтів (10%) виник рецидив, натомість нормальна функція наднирників зберігалася в 16 пацієнтів (62%). Очевидно, третини однієї функціональної надниркової залози досить для нормальної секреції глюко- та мінералокортикоїдів.
Синдром Кушинга
Гіперкортизолемія асоціюється з АГ приблизно у 80% дорослих і 50% дітей. Після лікування синдрому Кушинга приблизно 30% пацієнтів мають стійку АГ. У більшості дітей і підлітків нормалізація АТ відбувається протягом року; ймовірно, це залежить від ступеня та тривалості гіперкортизолемії до операції. Гіпертензія при синдромі Кушинга зумовлена такими механізмами: збільшення утворення ангіотензиногена в печінці; посилення серцевого викиду під впливом глюкокортикоїдів; зниження продукції простагландинів через пригнічення фосфоліпази А; підвищення резистентності до інсуліну; перенасичення активності 11β-гідроксистероїддегідрогенази з посиленням мінералокортикоїдного ефекту через стимуляцію мінералокортикоїдного рецептора. Для кращого розуміння наслідків порушення синтезу гормонів надниркових залоз див. рисунок 4.
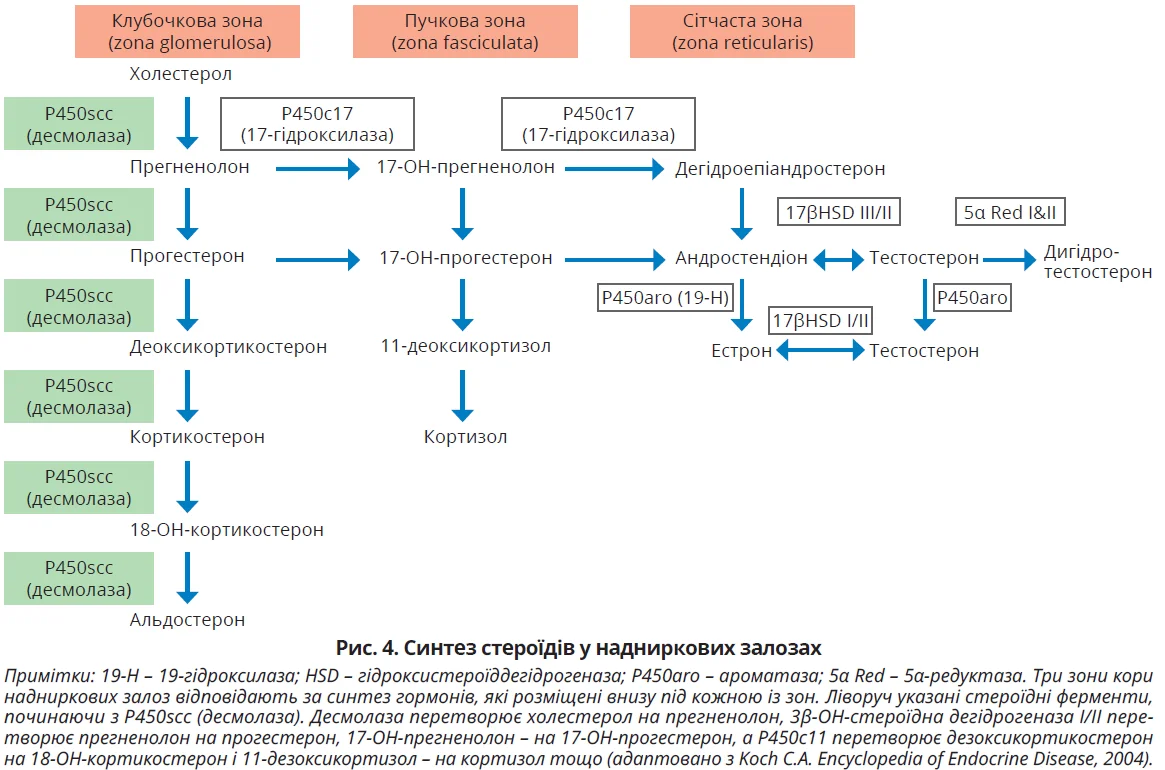
Скринінгові дослідження
Принаймні дворазове вимірювання 24-годинної екскреції вільного кортизолу із сечею; супресивний тест з 1 мг дексаметазону; визначення рівня кортизолу в слині (о 24:00); визначення добового ритму секреції кортизолу. В осіб без клінічних ознак синдрому Кушинга з інциденталомами надниркових залоз, виявлених методами візуалізації, субклінічний гіперкортицизм установлюється, якщо сироватковий рівень кортизолу о 8:00 становить >5 мкг/дл після введення 1 мг дексаметазону о 23:00 (чутливість – 58%, специфічність – 100%). Натомість рівень кортизолу <1,8 мкг/дл виключає синдром Кушинга.
Медикаментозне лікування
Терапія спрямована на усунення надлишку глюкокортикоїдів. Можливе використання тіазидних діуретиків. У разі гіпокаліємії краще призначати АМР (спіронолактон, еплеренон).
У проспективному рандомізованому дослідженні 45 пацієнтів із субклінічним гіперкортицизмом та інциденталомами надниркових залоз розподілено на дві групи: адреналектомії (n=23) та спостереження (n=22). Моніторинг включав контроль АТ, глікемії, ліпідного профілю, ожиріння й мінеральної щільності кісток. У групі адреналектомії покращення цукрового діабету спостерігалося в 62%, покращення АГ – у 67% пацієнтів; у групі спостереження відзначалося погіршення АТ, глікемічного контролю та ліпідного профілю.
Резистентність до глюкокортикоїдів (синдром Хрусоса)
Це автосомно-рецесивна чи автосомно-домінантна рідкісна спадкова хвороба, спричинена інактивувальними мутаціями гена глюкокортикоїдного рецептора з підвищенням кортизолу й АКТГ, але без клінічних ознак синдрому Кушинга. Постійне підвищення АКТГ призводить до стимуляції надниркових сполук із мінералокортикоїдною активністю (кортикостерон, DOC), що разом із гіперсекрецією кортизолу зумовлює стимуляцію мінералокортикоїдного рецептора та появу АГ.
У жінок через стимуляцію андрогенів (андростендіону, ДГЕА, 5-андростендіолу) може спостерігатися гірсутизм і олігоаменорея. Клінічно в дітей можуть бути неоднозначні геніталії та передчасне статеве дозрівання. Чоловіки можуть бути безплідними та/або мати олігоспермію. У жінок можливі акне, надмір волосся, дисменорея з олігоановуляцією та безпліддя. Лікування передбачає пригнічення секреції АКТГ високими дозами дексаметазону (1-3 мг/добу). Для контролю АТ використовують АМР (спіронолактон або еплеренон).
Вроджена гіперплазія наднирників
Дефіцит 11β-гідроксилази
Найпоширенішою причиною вродженої гіперплазії наднирників (ВГН) є дефіцит 21-гідроксилази. Власне АГ не розглядали як компонент цього синдрому, проте останні дані свідчать, що вона є поширенішою в цій когорті, ніж вважалося раніше.
Близько 5% усіх випадків ВГН зумовлено дефіцитом 11-β-гідроксилази – ферменту, відповідального за перетворення DOC на попередник альдостерону кортикостерон та 11-дезоксикортизолу на кортизол. Приблизно у 2/3 осіб із дефіцитом цього ферменту виникає моногенна АГ із низькими рівнями реніну й альдостерону, спричинена накопиченням 11-дезоксикортизолу та DOC. Іноді АГ виникає відразу після народження. Спосіб успадкування автосомно-рецесивний, зумовлений мутацією гена CYP11B1 у хромосомі 8. Через хронічно підвищений рівень кортикотропіну (АКТГ) і накопичення 17-OH прогестерону й андростендіону збільшується продукція андрогенів, що може призвести до внутрішньоутробної вірилізації та псевдогермафродитизму в жінок. У чоловіків можливі псевдораннє статеве дозрівання, низький зріст, іноді препубертатна гінекомастія. Зазвичай замісна терапія глюкокортикоїдами зменшує АГ; в окремих випадках проводиться двобічна адреналектомія.
Дефіцит 17-α-гідроксилази
Дефіцит 17-α-гідроксилази трапляється рідко та призводить до зниження утворення кортизолу й статевих стероїдів. Хронічне підвищення АКТГ спричиняє гіперпродукцію DOC і кортикостерону. Виникає АГ із низькими концентраціями альдостерону, пригніченням реніну, гіпокаліємією. У чоловіків спостерігається псевдогермафродитизм, у жінок – сексуальний інфантилізм і первинна аменорея. Іноді діагноз установлюється аж у період статевого дозрівання. Характерні низькі плазмові рівні надниркових андрогенів, кортизолу, 17-α-гідроксипрогестерону, альдостерону й низька активність реніну в плазмі; натомість рівні DOC, кортикостерон та 18-гідроксикортикостерону підвищені. Замісна терапія глюкокортикоїдами знижує АТ.
Пухлини, що продукують DOC
Це рідкісний тип пухлин надниркових залоз, які здебільшого є великими та злоякісними; разом із дезоксикортизоном можуть також виділяти андрогени й естрогени. У жінок можлива вірилізація, в чоловіків – фемінізація. Гіпокаліємія й АГ проявляються швидко; ренін і альдостерон часто знижені.
Явний надлишок мінералокортикоїдів
Зумовлюється дефіцитом ферменту 11-β-гідроксистероїддегідрогенази 2 типу (11-β-HSD2). Вроджений дефіцит – це автосомно-рецесивний розлад, уперше описаний New і співавт. у 1977 р.; спричиняється мутацією в гені 11β-HSD2, розташованому на хромосомі 16q22 (Wilson et al., 1995). Набутий дефіцит 11-β-HSD2 зумовлений пригніченням ферменту гліциризиновою кислотою, що міститься в солодці, жувальному тютюні та карбеноксолоні.
Фермент 11-β-HSD2 перетворює кортизол на неактивний кортизон у клітинах ниркових канальців, де коекспресується з мінералокортикоїдним рецептором. Кортизон не зв’язується з мінералокортикоїдними рецепторами, тоді як кортизол і альдостерон зв’язуються з ними з однаковою спорідненістю. Проте нормальна циркулювальна концентрація кортизолу в 100-1000 разів вища, ніж альдостерону. Дефект 11-β-HSD2 призводить до того, що для зв’язування з мінералокортикоїдним рецептором доступно більше кортизолу.
Часто спостерігаються затримка росту / низький зріст, АГ, гіпокаліємія, нецукровий нирковий діабет і нефрокальциноз, які виникають у дитинстві. Знижена активність 11-β-HSD2 може відігравати певну роль у патогенезі прееклампсії.
Діагноз установлюють шляхом вимірювання вільних некон’югованих стероїдів у сечі (співвідношення вільного кортизолу / вільного кортизону) та/або метаболітів стероїдів (тетрагідрокортизол + алотетрагідрокортизол/тетрагідрокортизон). Характерні низькі плазмові рівні реніну й альдостерону, гіпокаліємія, але нормальний плазмовий рівень кортизолу. Лікування включає спіронолактон, еплеренон, тріамтерен або амілорид. Трансплантація нирки є варіантом для пацієнтів із прогресивною нирковою недостатністю.
Конститутивна активація мінералокортикоїдних рецепторів (синдром Геллера)
Мутації рецептора мінералокортикоїдів призводять до початку АГ у віці до 20 років. Експерименти in vitro демонструють, що прогестерон і спіронолактон, які зазвичай є антагоністами рецептора мінералокортикоїдів, у разі синдрому Геллера стають агоністами. Відповідно, це мутації «посилення функції» в гені рецептора мінералокортикоїдів на хромосомі 4q31. Схема успадкування автосомно-домінантна.
Синдром Ліддла
Це автосомно-домінантний спадковий синдром, уперше описаний Liddle 1963 р. Зумовлений мутаціями «посилення функції» в генах, які кодують β- чи γ-субодиницю ниркового епітеліального натрієвого каналу, розташованого в хромосомі 16p13, що призводять до конститутивної активації ниркової реабсорбції натрію та подальшої гіперволемії.
Характеризується тяжкою АГ, гіпокаліємією й метаболічним алкалозом, низьким рівнем альдостерону та низькою активністю реніну в плазмі крові, але нормальним співвідношення кортизон/кортизол у добовій сечі. Обмеження солі й терапія тріамтереном покращують контроль АТ, спіронолактон неефективний.
Псевдогіпоальдостеронізм 2 типу (синдром Гордона)
Це рідкісний менделівський розлад з автосомно-домінантним успадкуванням, зумовлений мутаціями у WNK-кіназах WNK1 та WNK4 на хромосомах 12 і 17 відповідно. За клінічний фенотип відповідає активація мутацій у чутливому до амілориду натрієвому каналі дистального відділу ниркових канальців. Поширеність синдрому Гордона невідома, оскільки багато пацієнтів залишаються без діагнозу. У родинах із діагностованою хворобою (походження з Австралії чи США) спостерігаються АГ із низьким рівнем реніну, гіперкаліємія, метаболічний ацидоз, нормальна функція нирок, низький/нормальний рівень альдостерону.
Механізмом АГ є підвищення реабсорбції солі в нирках; гіперкаліємія виникає внаслідок зниження ниркової екскреції калію, незважаючи на нормальну клубочкову фільтрацію та секрецію альдостерону. Знижена ниркова секреція калію робить цей стан схожим на стан дефіциту альдостерону, тому вживається термін «псевдогіпоальдостеронізм». Притаманна залежність від хлориду – порушення корегує інфузія хлориду натрію, а не гідрокарбонату натрію. Встановлено також, що всі прояви покращуються суворим обмеженням харчової солі. Отже, для контролю АГ при цьому синдромі показано суворе обмеження солі в їжі та тіазидні діуретики.
Інсулінорезистентність
Метаболічний синдром характеризується АГ, абдомінальним/вісцеральним ожирінням, дисліпідемією та резистентністю до інсуліну. Принаймні 24% дорослих у США відповідають критеріям діагнозу метаболічного синдрому. Інсулінорезистентність значною мірою пов’язана з АГ у латиноамериканців, може спричинити судинну дисфункцію та часто спостерігається в пацієнтів з есенціальною АГ.
Цікаво, що не всі пацієнти з інсулінорезистентністю мають ожиріння. Проте надмірна маса тіла становить близько 70% ризику есенціальної АГ, а також підвищує ризик термінальної стадії ниркової недостатності. Інсулін здатний прямо стимулювати кальцієву помпу в чутливих до нього тканинах, що призводить до втрати внутрішньоклітинного кальцію. Підвищені цитозольні концентрації кальцію в адипоцитах можуть спричиняти інсулінорезистентність. У клітинах, резистентних до інсуліну, зменшується інсулін-індукована втрата кальцію. Збільшення внутрішньоклітинного кальцію в клітинах гладенької мускулатури судин призводить до активнішої реакції на вазоконстриктори та підвищення АТ.
Серед інших механізмів, які можуть пояснити зв’язок інсулінорезистентності й АГ, – затримка натрію та посилена активність адренергічної нервової системи. При ожирінні надмірне утворення адипокінів жировою тканиною впливає на численні функції, зокрема й чутливість до інсуліну, АТ, ліпідний обмін.
Первинний гіперпаратиреоз
Рівень паратгормону відповідає концентрації кальцію в сироватці крові; в пацієнтів з АГ паратгормон зазвичай у межах норми. Проте пацієнти з есенціальною АГ виділяють більше кальцію порівняно з людьми з нормальним тиском, що свідчить про посилення функції паращитоподібних залоз. Інфузія паратгормону супроводжується судинорозширювальним ефектом, але тривала інфузія підвищує АТ у здорових людей. Високе споживання кальцію може знизити АТ, але гіперкальціємія асоціюється зі збільшенням частоти АГ.
У 40% пацієнтів із первинним гіперпаратиреозом спостерігається АГ. Її точний механізм не з’ясований, можливо, пов’язаний із посиленням жорсткості артерій, аномаліями сонних артерій, більшою варіабельністю САТ і ендотеліальною дисфункцією.
У пацієнтів із безсимптомним перебігом паратиреоїдектомія не показала жодних переваг щодо АТ чи якості життя порівняно з медикаментозним лікуванням. Проте тяжка АГ виліковується або краще контролюється після паратиреоїдектомії. Крім того, паратиреоїдектомія може знизити ризик серцево-судинних захворювань унаслідок зниження загального холестерину. Іншими показаннями до паратиреоїдектомії є ураження скелета та/або нефрокальциноз, підтверджений методами візуалізації.
Гіпертиреоз
Механізмами АГ за гіпертиреозу та тиреотоксикозу є збільшення САТ унаслідок тахікардії, зниження системного судинного опору, підвищення серцевого викиду й ударного об’єму. Близько третини пацієнтів із гіпертиреозом мають АГ, яка часто зникає після досягнення еутиреозу. Субклінічний гіпертиреоз може спричиняти гіпертрофію лівого шлуночка, а отже, призводити до АГ, хоча це до кінця не встановлено.
Гіпотиреоз
Пацієнти з гіпотиреозом мають порушення функції ендотелію, підвищений системний судинний опір, збільшення позаклітинного об’єму та підвищення ДАТ, а також вищий середній 24-годинний САТ і більшу варіабельність АТ за 24-годинного амбулаторного моніторингу. У 32% пацієнтів з АГ і гіпотиреозом замісна терапія тироксином сприяє зниженню ДАТ до ≤90 мм рт. ст. Гіпотиреоз також може спричиняти об’ємозалежне підвищення АТ із низькою концентрацією реніну в плазмі крові. Субклінічний гіпотиреоз не обов’язково пов’язаний з АГ.
Акромегалія
Поширеність АГ у пацієнтів із надлишком гормону росту становить ≈46%, що частіше, ніж у загальній популяції. Гормон росту має антинатрійуретичну дію, що призводить до затримки натрію й гіперволемії. Збільшений систолічний викид і тахікардія внаслідок гіперкінетичного синдрому здатні призвести до застійної серцевої недостатності. Високі показники АТ можуть також асоціюватися з порушеннями обміну глюкози або діабетом. Схоже, що в патогенезі АГ у пацієнтів із надлишком гормону росту задіяна активація РААС.
Притаманні акромегалії гіперліпідемія, АГ, цукровий діабет і кардіоміопатія можуть покращитися навіть за часткового біохімічного контролю надлишку гормону росту. Проте в деяких пацієнтів АГ та діабет можуть зберігатися після спроб біохімічного лікування або під час ремісії.
Інші ендокринні стани, що спричиняють АГ
З’являється дедалі більше доказів того, що дефіцит вітаміну D може бути пов’язаний з АГ та підвищеним серцево-судинним ризиком.
Потенційними механізмами в цьому випадку є одночасна резистентність до інсуліну та пряма дія вітаміну D через РААС (рис. 5).
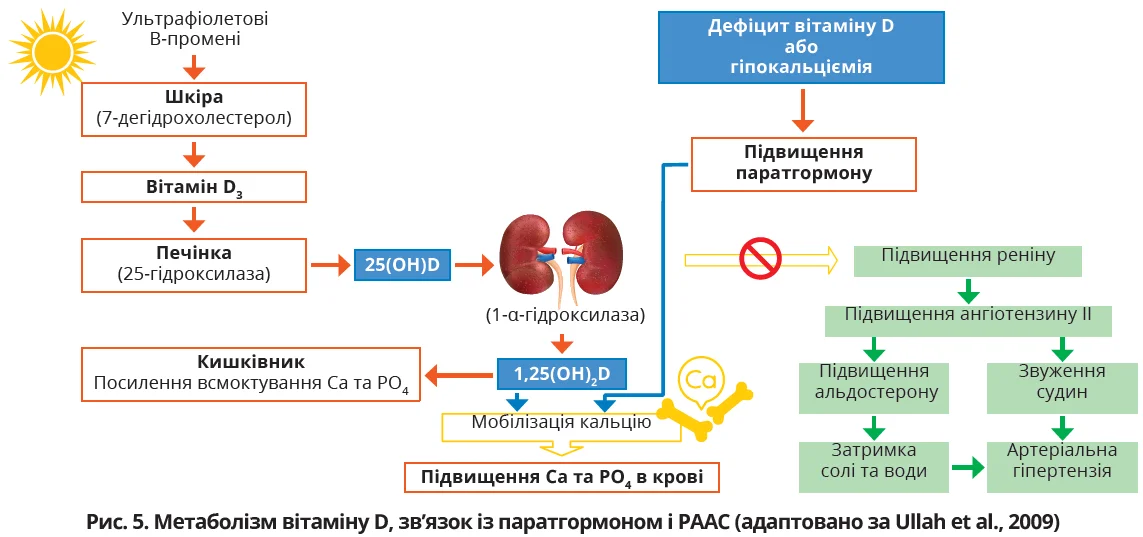
Дефіцит тестостерону часто виявляють у людей з ожирінням і цукровим діабетом або метаболічним синдромом. Замісна терапія іноді корисна не лише в корекції симптомів, пов’язаних із дефіцитом андрогенів (низьке лібідо, погана ерекція, втома), але й у зв’язку з АТ і метаболічним профілем. Подібним чином люди з дефіцитом гормону росту можуть бути схильні до АГ, здебільшого через те, що склад їхнього тіла є більш «жирним» і «запаленим» порівняно з особами з адекватною кількістю гормону росту (оцінюється за рівнями IGF-1 у сироватці крові відповідно до статі та віку). Ключовими в таких пацієнтів будуть збільшення фізичної активності й індивідуальна замісна терапія гормоном росту до нормалізації IGF-1. Для пацієнтів з ожирінням, які бажають суттєво змінити спосіб життя для схуднення, можна на певний час призначити фентермін, топірамат, ліраглутид, лоркасерин, орлістат або налтрексон/бупропіон разом з уведенням гормону росту.
Індивідуальна тканинозалежна чутливість глюкокортикоїдного рецептора та дії ендогенних глюкокортикоїдів можуть відігравати важливу роль у виникненні АГ, ожиріння й цукрового діабету.
Література
Koch C., Papadopoulou-Marketou N., Chrousos G.P. Overview of endocrine hypertension [Updated 2020 Feb 4]. In: Feingold K.R., Anawalt B., Blackman M.R., et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000. Available at: https://www.ncbi.nlm.nih.gov/books/NBK278980.